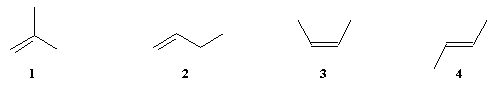
Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
LES
ALCÈNESGénéralités
Nomenclature
On appelle alcènes, les hydrocarbures insaturés non cycliques de
formule CnH2n.
La chaîne principale est celle qui comporte le plus grand nombre de liaisons
et, le cas échéant, le plus grand nombre d'atomes de carbone. Le nom du composé
dérive de celui de l'alcane en remplaçant la terminaison ane par ène.
La position de la double liaison est indiquée par le numéro de l'atome de
carbone doublement lié qui
|
1 |
2 |
3 |
4 |
|
Méthylpropène |
But-1-ène |
(Z)-But-2-ène |
(E)-But-2-ène |
Les composés éthyléniques constituent un ensemble plus large que les alcènes, de composés comportant une double liaison carbone-carbone. Les hydrocarbures cycliques ayant une double liaison ou cyclènes appartiennent à cette catégorie. On notera qu'une double liaison éthylénique ne constitue pas un groupe fonctionnel.
|
|
L'éthène est l'alcène le plus important sur le plan industriel. Il est produit par craquage d'hydrocarbures.
|
|
|
Le propène est le deuxième composé éthylénique le plus important comme produit de base de l'industrie chimique. La part la plus importante sert à la synthèse du polypropène (polypropylène). Le reste permet la préparation des composés suivants :
|
Cyclènes
Les cyclènes sont des hydrocarbures de formule CnH2n-2.
Les cycles moyens possédant 5 ou 6 atomes de carbone possèdent une réactivité
comparable à celle des alcènes. Certains cyclènes possèdent un plan de
chiralité. Le trans-cyclooctène
est dédoublable en deux énantiomères.
Diènes, polyènes
Les diènes sont des composés éthyléniques qui possèdent deux liaisons
doubles. A côté des diènes dont les doubles liaisons sont éloignées
et qui se comportent comme des éthyléniques ordinaires, on distingue :
Propriétés physiques
Structure de l'éthène
La simple connaissance de la formule brute C2H4 est en
faveur d'une liaison double entre les atomes de carbone. Le tableau
suivant rassemble des données expérimentales relatives à l'éthane et à l'éthène.
|
Liaison |
Longueur (nm) |
Energie (kJ.mol-1) |
|
C-C |
0,154 |
347 |
|
C=C |
0,133 |
606 |
La liaison entre les atomes de carbone dans l'éthène est plus courte et plus énergétique que dans l'éthane. Notons que l'énergie de dissociation de cette liaison double est plus petite que le double de celle d'une liaison simple.
Géométrie et isomérie
Les atomes impliqués dans la liaison double ont une géométrie localement
plane. L'existence de la double liaison chez les alcènes bloque la libre
rotation des groupes qui lui sont attachés. Elle est à l'origine de la diastéréoisomérie
Z, E.
Constantes physiques
Les alcènes ont des propriétés physiques assez comparables à celles des
alcanes. Il est intéressant de noter la différence de propriétés entre des
alcènes stéréoisomères comme l'illustre le cas des but-2-ène.
|
Composé |
Moment dipolaire (D) |
Température d'ébullition (°C) |
|
(Z)-But-2-ène |
0,4 |
4 |
|
(E)-But-2-ène |
0 |
1 |
Orbitales moléculaires
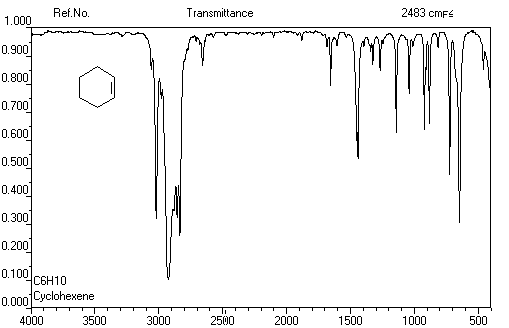
Niveaux d'énergie Spectroscopie Spectroscopie infrarouge Composé s (cm-1) 890 915 & 995 970 Exemple : Spectre infrarouge du cyclohexène
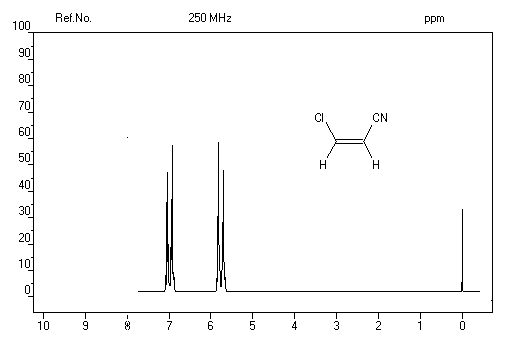
Composé Constante de couplage 1 16 Exemple : spectre RMN du 1-chloro-2-cyano-éthène Hydrogénation Conditions expérimentales Comparaison de la stabilité des alcènes -118,9 -
(-114,6) = - 4,3
kJ.mol-1
Cette différence de stabilité peut s'interpréter par des contraintes stériques
entre les groupes méthyle qui sont plus importantes dans l'isomère Z que dans
l'isomère E. Stéréochimie Dans des conditions comparables, l'addition de H2 sur le 3,4-diméthylhex-3-ène
de configuration E fournit de façon quasi-exclusive le mélange
racémique des énantiomères (3R,4R) et (3S,4S) du 3,4-diméthylhexane.
Elle est aussi, fortement stéréosélective et la géométrie de l'addition est
également syn.
Deux substrats stéréoisomères fournissent dans les mêmes conditions des
produits diastéréoisomères. La réaction d'hydrogénation est donc diastéréospécifique.
La stéréochimie de l'addition est syn. Catalyse hétérogène Mécanisme et cinétique Le mécanisme précédent explique l'influence sur la vitesse de réaction du
nombre et de la position des substituants autour de la double liaison.
On peut mettre à profit les différences de vitesse d'hydrogénation pour réaliser
des hydrogénations sélectives. Dans l'exemple ci-dessous, l'hydrogénation
de la double liaison la plus accessible du limonène est réalisée sélectivement
sous contrôle cinétique :
Catalyse homogène C'est un catalyseur chimiosélectif particulièrement
efficace permettant l'hydrogénation en phase homogène des doubles
liaisons éthyléniques Halogénation
Résultats expérimentaux On lui préfère actuellement le procédé d'oxychloration dans lequel l'élément
chlore est apporté par le mélange O2 / HCl. Les composés éthyléniques additionnent facilement Cl2 et Br2
pour conduire à des dérivés dihalogénés vicinaux. F2 très
oxydant détruit la molécule. Avec I2 peu réactif, la réaction
conduit à un équilibre défavorable au produit. La réaction s'effectue par
union directe dans les conditions normales de température et de pression. Les
solvants utilisés sont CH2Cl2, CHCl3, CCl4,
AcOH. Test analytique des doubles liaisons éthyléniques I II III Stéréochimie Dans les mêmes conditions, la réaction effectuée sur le (E)-3,4-diméthylhex-3-ène,
fournit essentiellement le mélange des énantiomères de configuration
absolue (3R, 4R) et (3S, 4S) La réaction de bromation est par conséquent diastéréospécifique
de stéréochimie anti.
Mécanisme Ions bromonium L'intervention d'ions bromonium dans les réactions de bromation a
pu, dans certains cas, être démontrée de façon directe.
Les considérations de symétrie précédentes sont très importantes pour
l'application de la théorie des orbitales moléculaires. Une simplification
importante intervient pour les molécules qui comme l'éthène sont planes. La réflexion
par rapport au plan de la molécule (xOy) permet de séparer les orbitales moléculaires
en deux groupes :
Ces dénominations simplificatrices proviennent d'une extension de la
terminologie valable en toute rigueur pour les molécules linéaires.
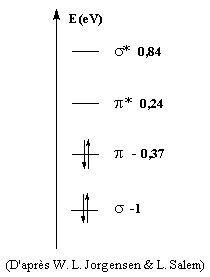
L'étagement des niveaux d'énergie moléculaires peut être obtenu
par le calcul.
Le diagramme complet des orbitales moléculaires de l'éthène
comporte 12 niveaux. L'image de gauche représente un diagramme
simplifié ne comportant que les orbitales de symétrie p
et celles de symétrie s respectivement
liante et anti-liante pour tous les atomes de la molécule.
La vibration de valence de la double liaison absorbe vers 1650 cm-1.
Les fréquences d'absorption des vibrations de déformation des liaisons C-H
sont également précieuses pour identifier les structures. Quelques absorptions
caractéristiques sont données ci-dessous.
d = 5,6 ppm. D'une façon générale, les atomes
d'hydrogène liés à la double liaison appelés hydrogènes vinyliques,
ont des déplacements chimiques assez élevés.

La réaction entre le dihydrogène H2 et un composé éthylénique
est une réaction d'addition. Elle n'est réalisable qu'à une vitesse raisonnable qu'avec l'aide d'un
catalyseur. D'un point de vue
thermodynamique, elle est quasi-totale dans les conditions normales de température
et de pression. Le rendement est de l'ordre de 90 %.
Puisqu'un catalyseur accélère aussi bien la réaction directe que la réaction
inverse, la déshydrogénation d'un hydrocarbure saturé est également
envisageable mais elle constitue surtout une méthode industrielle en raison de
son manque de sélectivité.
La réaction d'hydrogénation peut être mise à profit pour évaluer la différence
de stabilité entre alcènes stéréoisomères.
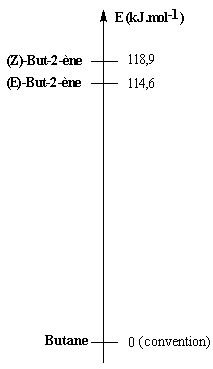
L'hydrogénation du (Z)-but-2-ène dégage une énergie
de -118,9 kJ.mol-1.
Celle du (E)-but-2-ène dégage -114,6 kJ.mol-1.
On en déduit l'enthalpie de la réaction suivante :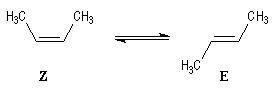
Le stéréoisomère E est donc plus stable que le stéréoisomère
Z
 E
E
 Z
Z
L'addition de H2 sur le 3,4-diméthylhex-3-ène de configuration Z
fournit de façon quasi-exclusive le (3R,4S)-3,4-diméthylhexane achiral.
Le (3R,4R)-3,4-diméthylhexane et son énantiomère le (3S,4S)-3,4-diméthylhexane,
tous trois diastéréoisomères du précédent ne sont pas obtenus. La réaction
est donc fortement diastéréosélective.
Les atomes d'hydrogène se sont fixés du même côté de la double liaison. La
stéréochimie de l'addition est syn.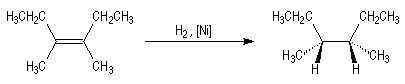

L'hydrogénation catalytique utilisant un métal de transition a été découverte
en 1912. Sabatier utilisait du nickel Ni préparé par réduction de NiO par le
dihydrogène.
Le caractère syn de l'addition peut être interprété grâce au modèle
suivant dans lequel les réactifs adsorbés à la surface du catalyseur sont
dans une disposition géométrique favorable pour la réaction d'addition.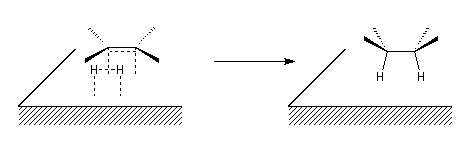
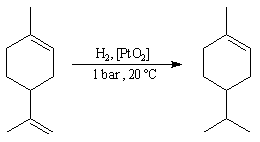
L'hydrogénation peut aussi être réalisée en milieu homogène en utilisant un
catalyseur soluble comme le catalyseur de Wilkinson.
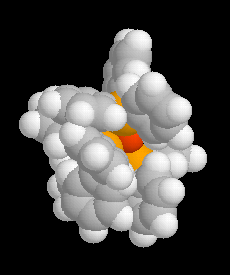
L'image de gauche représente un modèle compact de [RhCl(PPh3)3].
Ce composé a été découvert en 1965. On l'obtient par réaction
entre la triphénylphosphine
et le trichlorure de rhodium dans l'éthanol.
![]()
![]()
L'une des premières synthèse industrielle du dichloroéthane consistait en une réaction
d'addition entre l'éthène et le dichlore.
La réaction de bromation peut être utilisée comme test des doubles liaisons
éthyléniques. Sur la photographie I l'erlenmeyer contient une
solution de dibrome Br2 dans CCl4 (on observe les
vapeurs de Br2 qui s'échappent de la solution). L'ajout de
quelques gouttes d'hex-1-ène provoque une décoloration rapide de la solution
(photos II et III).



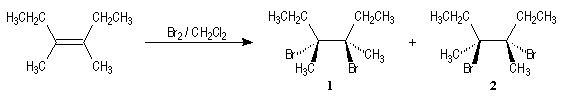
L'addition de Br2 sur le (E)-3,4-diméthylhex-3-ène fournit quasi
exclusivement le (3R,4S)-dibromo-3,4-diméthylhexane.
Puisqu'on obtient un seul stéréoisomère parmi plusieurs pouvant se former a
priori, la réaction de bromation est fortement diastéréosélective.
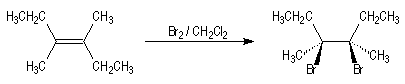
Il s'agit d'un mécanisme par stades :
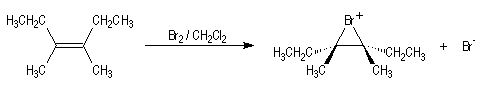
Ce mécanisme rend compte de la stéréospécificité anti de la réaction.
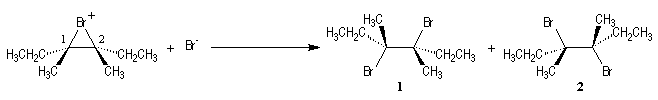
Les ions bromonium sont des intermédiaires très réactifs qui ne sont généralement
pas isolés.
Protection des doubles liaisons éthyléniques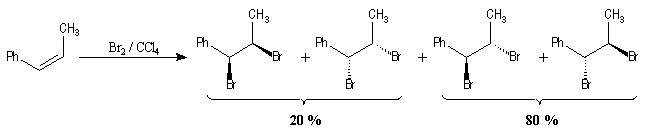
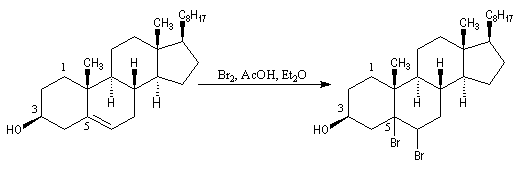
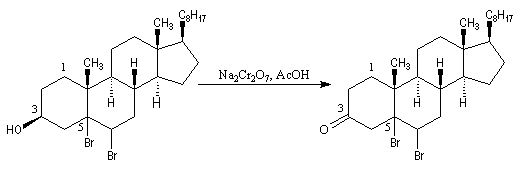
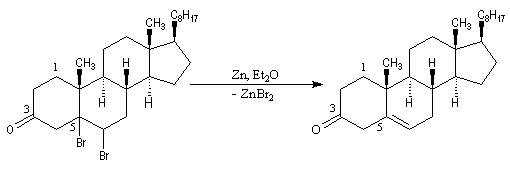
L'oxydation du cholestérol
en milieu acide, pose le problème de l'isomérisation de la cholest-5-ène-3-one
en cholest-4-ène-3-one, une cétone
conjuguée plus stable. Une solution consiste à bloquer la double liaison éthylénique.
Addition des hydracides
Résultats expérimentaux
La réaction entre HBr dissous dans l'acide éthanoïque et le méthylpropène
à 20 °C donne essentiellement du 2-bromo-2-méthypropane (1) et
seulement des traces de 1-bromo-2-méthylpropane (2).

Mécanisme
Il s'agit d'un mécanisme par stades :
![]()
![]()
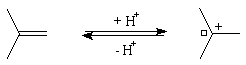
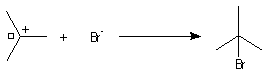
Remarque : une addition anti de HBr est parfois observée. Elle s'interprète par la formation d'un complexe entre le composé éthylénique et l'hydracide.
Hydratation
Résultats expérimentaux
L'addition d'eau sur un composé éthylénique s'appelle hydratation. Elle
conduit à la formation d'un alcool. Avec les alcènes substitués qui forment facilement des carbocations, il
suffit d'utiliser un acide dilué. L'ion H+ est alors un catalyseur
de la réaction :
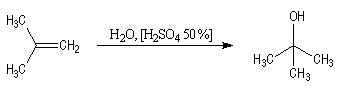
Avec les alcènes moins réactifs, on utilise de l'acide sulfurique concentré. Il y a formation d'un sulfate d'alkyle intermédiaire.
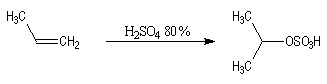
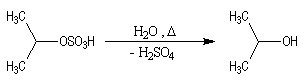
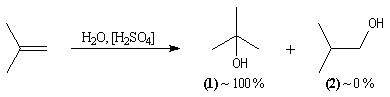
Mécanisme
Il ressemble beaucoup à celui décrit pour l'addition de HBr. Il s'agit d'un
mécanisme par stades. On peut regarder cette réaction comme la réaction
inverse de la déshydratation
des alcools.
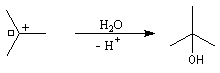
Règle de Markovnikov
Enoncé historique
Cette règle a été énoncée en 1868. Il
s'agissait à l'origine d'une règle empirique qui concernait l'addition des
hydracides halogénés sur les alcènes dissymétriques. Markovnikov avait
reconnu le caractère très régiosélectif de cette addition. La règle
permet de prévoir l'atome de carbone sur lequel se fixe l'hydrogène de
l'hydracide :
Lors de l'addition d'un hydracide sur un alcène dissymétrique l'atome d'hydrogène se fixe sur l'atome de carbone le moins substitué.
Exemple:
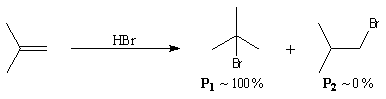
Généralisation de la règle de Markovnikov
L'expérience montre que de nombreuses additions de réactifs dissymétriques
s'effectuent sous contrôle cinétique par un mécanisme ionique impliquant un
carbocation ou un ion positif ponté. La règle de Markovnikov peut être généralisée
:
Lors de l'addition d'un réactif Ad -- Hd+ sur un composé éthylénique dissymétrique, Ad - se fixe préférentiellement sur l'atome de carbone qui stabilise le mieux une charge positive.
Lorsque le composé éthylénique est un alcène, l'atome de carbone qui stabilise le mieux une charge positive est celui qui est substitué par le plus grand nombre de groupes alkyles. Pour des alcènes fonctionnalisés, la situation est plus délicate et il faut examiner chaque cas.
Hydroboration
Résultats expérimentaux
Le monoborane BH3 est un composé déficient en électrons par
rapport à l'octet. A la température ordinaire, il se dimérise pour conduire
au diborane B2H6. Mais en présence de THF meilleure
base de Lewis que H, il y a formation d'un complexe entre le monoborane et l'éther.
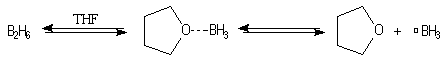
L'entité réactive est le monoborane BH3 qui résulte de la
faible dissociation du complexe.
La réaction est régiosélective. Le bore se fixe sur l'atome de
carbone le moins substitué de la double liaison.
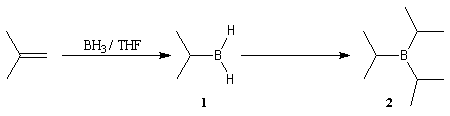
Mécanisme de l'hydroboration
Il s'agit d'une cycloaddition entre une liaison bore-hydrogène et une
liaison carbone-carbone. Le bore se fixe de façon régiosélective sur
l'atome de carbone le moins substitué. La raison principale de cette régiosélectivité
semble être d'origine stérique. Elle fait sans doute aussi intervenir des
facteurs électriques. On remarquera à cet égard que le bore est moins électronégatif
que l'hydrogène. Le schéma simplifié est le suivant :
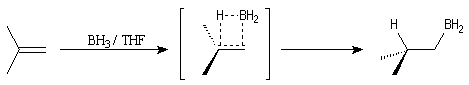
Additions radicalaires
Résultats expérimentaux
En étudiant l'addition de bromure d'hydrogène sous rayonnement ultraviolet
le chimiste américain d'origine russe Kharasch a mis en évidence une
addition de régiosélectivité inverse de celle observée pour l'addition
ionique. L'orientation de l'addition est pour cette raison qualifiée d'anti-Markovnikov.
Mécanisme
Le mécanisme de cette réaction a été élucidé par Kharasch et Mayo en 1930. Il s'agit d'une réaction en chaîne.
![]()

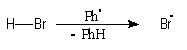
![]()
![]()
![]()
Régiosélectivité Hydroformylation
La régiosélectivité s'interprète par un contrôle cinétique de la réaction.
L'étape cinétiquement déterminante est la formation du radical le plus
stable.
Seul le bromure d'hydrogène est capable de réagir avec les composés éthyléniques
selon ce mécanisme.
Résultats expérimentaux ou procédé oxo Epoxydation
L'hydroformylation est une réaction industrielle découverte en 1938. Il permet l'insertion d'oxygène dans une chaîne carbonée.
Le bilan est le
suivant :
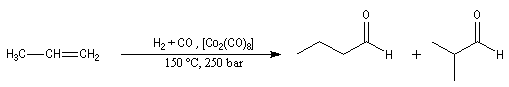
Préparation des époxydes
La réaction entre un peroxyacide et un composé éthylénique conduit à
la formation d'un époxyde. L'acide métachloroperoxybenzoïque m-CPBA a été
longtemps le réactif de peroxydation le plus utilisé. A partir du (Z)-but-2-ène
comme substrat, on obtient le (cis)-2,3-diméthyloxacyclopropane :
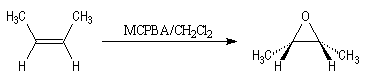
Réalisée à partir du diastéréoisomère de stéréochimie E, la réaction conduit à un mélange racémique d'époxydes énantiomères.
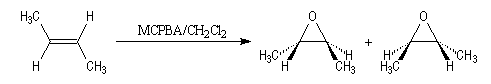
La réaction est stéréospécifique de stéréochimie syn.
|
|
L'acide métachloroperoxybenzoïque m-CPBA fut très utilisé comme réactif permettant l'époxydation des doubles liaisons éthyléniques. Comme tous les composés organiques comportant une liaison peroxo fragile, il est peu stable. Ce réactif est à l'origine de plusieurs accidents. Il est le plus souvent remplacé de nos jours par le monoperoxophtalate de magnésium MMPP.
|
Dihydroxylation
Utilisation du permanganate de potassium
La réaction s'effectue avec une solution de permanganate diluée, en milieu
neutre à une température voisine de 0 °C. Un ester permanganique se forme
intermédiairement. Des expériences utilisant de l'oxygène 18 ont montré
que l'addition s'effectue avec une stéréochimie syn. L'hydrolyse de
cet ester fournit le diol cis.
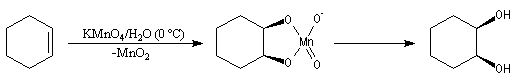
|
|
L'oxydation d'un composé éthylénique en diol par le permanganate de potassium doit être réalisée avec une solution diluée, en milieu neutre dans un bain de glace. A température plus élevée, l'oxydation provoque une rupture de la chaîne carbonée et conduit à la formation de composés carbonylés. Les aldéhydes éventuels sont oxydés en acides carboxyliques |
Utilisation du tétraoxyde d'osmium
A cause de son coût très élevé et de son caractère toxique, le tétraoxyde
d'osmium est utilisé en quantité catalytique en présence de peroxyde
d'hydrogène H2O2.
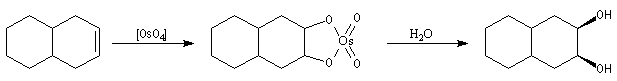
Ozonation, ozonolyse
Résultats expérimentaux
La réaction entre l'ozone et un composé éthylénique s'appelle ozonation.
Elle est réalisée à basse température et fournit un ozonide.
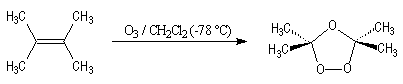
|
|
Le chimiste allemand C. F.
Shönbein découvrit l' ozone O3 en
1839. C'est un allotrope du dioxygène. L'ozone est présent à l'état
naturel dans la haute atmosphère de la terre. Au laboratoire on le prépare
en faisant traverser le dioxygène O2 par une effluve électrique
dans un ozoniseur. A la température ordinaire l'ozone est un gaz de
couleur bleue possédant une odeur piquante qui lui a valu son nom. L'ozone se liquéfie à -112
°C pour donner un liquide bleu explosif. Conformément au prévisions de la méthode VSEPR, la molécule d'ozone a la forme d'un V avec un angle entre les liaisons de 116,8°. |
Il n'est pas possible de décrire la molécule d'ozone par une formule de Lewis unique. On peut rendre compte de sa structure en utilisant les formes mésomères suivantes :
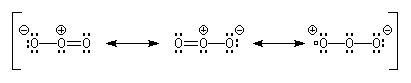
Mécanisme
La réaction
entre O3 et la double liaison éthylénique est une réaction péricyclique.
Elle conduit dans un premier temps à un intermédiaire particulièrement
instable : le molozonide M. Ce dernier se fragmente spontanément
pour conduire à l'ozonide O. Le schéma simplifié est le suivant :
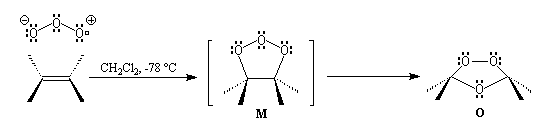
Les ozonides sont rarement isolés du milieu réactionnel mais sont mis à réagir in situ avec le réactif souhaité. Les ozonides sont des composés explosifs en raison de la fragilité de la liaison peroxo. La réaction doit être réalisée à basse température.
Ozonolyse
L'une des principales réactions des ozonides est leur coupure en composés
carbonylés. L'hydrolyse en milieu non réducteur fournit du peroxyde d'hydrogène
comme sous-produit qui oxyde les aldéhydes éventuellement formés en
acides carboxyliques ;
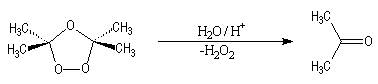
Recherche de structure
Depuis sa découverte , la réaction a été
largement utilisée pour localiser la position des doubles éthyléniques dans
les molécules organiques.
Supposons que l'ozonolyse d'un alcène fournisse uniquement de la butanone et
du méthanal. On en déduit la structure de cet alcène :
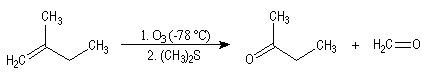
Utilisation en synthèse
L'ozonolyse est aussi utilisée pour préparer des composés carbonylés ou
dicarbonylés. La réaction suivante est une étape de la synthèse d'une phéromone
d'insecte. Le composé dicarbonylé est obtenu par coupure du méthylcycloheptène.
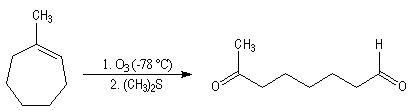
Oxydation en aldéhyde
Procédé Wacker
L'oxydation de l'éthylène par le dioxygène de l'air permet la préparation
industrielle de l'éthanal. Le procédé, mis au point en 1953 porte le nom de procédé Wacker. La réaction
fait intervenir un cycle catalytique complexe. Les principales étapes
sont les suivantes :
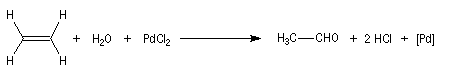
![]()
![]()