Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
LES ALCOOLS
Introduction
Nomenclature
On appelle alcool un composé dans lequel un groupe
hydroxyle -OH est lié à un atome de carbone
saturé.
La chaîne principale est la chaîne la plus longue qui porte le groupe -OH. La numérotation de la chaîne est choisie de façon que le groupe - OH ait le numéro le plus petit. Le nom de l’alcool est formé en ajoutant le suffixe ol au nom de l’hydrocarbure possèdant le même nombre d’atomes de carbone que la chaîne principale.
Les énols, composés dans lesquels le groupe -OH est lié à un atome de carbone insaturé ou les phénols dans lesquels ce groupe est lié à un cycle aromatique, ne sont pas des alcools.
Méthanol CH3OH
![]()
Ethanol C2H5OH
.![]()
Cyclohexanol C6H11OH
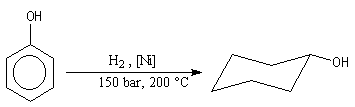
Propriétés physiques
Températures de changement d’état
Le tableau suivant regroupe les températures de changement d'état de quelques
alcools courants.
|
Nom de l’alcool |
TF (°C) |
TE (°C) |
Densité d |
|
Méthanol |
-97 |
64,7 |
0,792 |
|
Ethanol |
-114 |
78,3 |
0,789 |
|
Propan-1-ol |
-126 |
97,2 |
0,804 |
|
Propan-2-ol |
-88 |
82,3 |
0,786 |
|
Butan-1-ol |
-90 |
117,7 |
0,810 |
|
2-Méthylpropan-2-ol |
2 |
82,5 |
0,789 |
|
Hexan-1-ol |
-52 |
155,8 |
0,820 |
|
Dodécanol |
24 |
259 |
0,831 |
Spectroscopie
Spectroscopie infrarouge
Le spectre suivant est celui de l'hexan-1-ol. Il est typique du spectre
infrarouge d'un alcool pur.

Lorsque l'alcool est pur, on observe une bande large dans la partie gauche du spectre :
3200 cm-1 < s < 3400 cm-1
Elle correspond à la vibration de valence des liaison O-H associées par liaison H intermoléculaire.
Réactions d'oxydo-réduction
Réactifs usuels impliquant des éléments de transition
|
|
Le dichromate de potassium est un solide de couleur orange. Il est dissous dans une solution d'acide sulfurique. Le volume est complété par de l'eau distillée. Les composés du Cr (VI) sont dangereux. Il présentent malheureusement la propriété d'induire certains cancers. Comme l'absorption par voie cutanée constitue le risque principal, il est indispensable d'utiliser des gants pour manipuler ces composés. On peut effectuer le dosage de l'éthanol en le faisant réagir avec un volume connu de solution titrée de dichromate de potassium en excès ce qui permet de rendre la réaction d'oxydation quantitative. Le dichromate restant est réduit par une solution titrée de sel de Mohr. |
Influence de la classe de l'alcool
|
|
Les deux tubes de gauche contiennent respectivement du butan-1-ol et une solution de dichromate de potassium dans l'acide sulfurique. Dans le tube de droite, on a introduit une petite quantité d'alcool dans la solution de dichromate de potassium. Une coloration bleu-vert se développe qui témoigne de la réduction des ions Cr2O72- en ions Cr3+. |
|
|
Dans le tube où s'est déroulée l'oxydation, on a ajouté une petite quantité de pentane. Après agitation, le butanal formé par oxydation de l'alcool se concentre dans cette phase organique (phase supérieure). Quelques mL de la phase organique surnageante sont ajoutés dans deux autres tubes : le premier contient une solution de réactif de Schiff qui vire au rose. le second contient une solution de 2,4-DNPH dans laquelle on observe un précipité de 2,4-dinitrophénylhydrazone. |
Les différences de comportement des alcools vis à vis de l'oxydation sont très nettes suivant la classe à laquelle ils appartiennent. La présence d'un atome d'hydrogène sur l'atome fonctionnel est indispensable pour que l'alcool soit oxydé. Les alcools tertiaires, ne sont pas oxydés.
Alcools secondaires
Le réactif de Jones est un réactif souvent employé (on dissout 26,72 g de
CrO3 dans 23 mL de H2SO4
concentré puis on étend avec de l'eau jusqu'à un volume de 100 mL).
L'oxydation des alcools secondaires conduit aux cétones. Exemple:
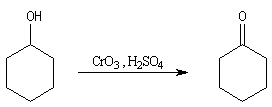
Alcools primaires
L'oxydation des alcools primaires conduit dans un premier temps à l'aldéhyde.
Mais les aldéhydes étant des réducteurs forts il faut prendre des précautions
spéciales pour s'arrêter à ce stade. Un certain nombre de techniques sont
utilisables. Les aldéhydes sont généralement plus volatils que les alcools parents car
ils ne forment pas de liaison H. Il est parfois possible de distiller l'aldéhyde
au fur et à mesure de sa formation ce qui a pour effet de supprimer le contact
avec l'oxydant. On peut ainsi préparer le butanal par oxydation du butan-1-ol avec Na2Cr2O7
en présence d'acide sulfurique. Le rendement n'est cependant pas très bon.
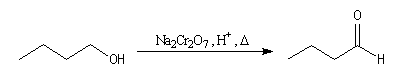
Une autre technique d'isolement est utilisée dans le test des alcools décrit plus haut. La méthode consiste à utiliser deux phases non miscibles : par exemple eau et pentane. Au fur et à mesure de sa formation, le butanal plus soluble dans le pentane que dans l'eau est extrait du milieu aqueux ce qui, la encore évite le contact avec le réactif d'oxydation.
Le réactif de Collins CrO3 (C5H5N)2 est également souvent utilisé dans l'oxydation des alcools primaires en aldéhydes pour des raisons analogues.
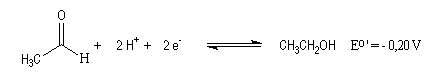
Notons que l'oxydation d'un alcool nécessite une base pour arracher un atome d'hydrogène de l'alcool.
Oxydations par le dioxygène
La synthèse du méthanal
s'opère par oxydation du méthanol par l'oxygène de l'air.
![]()
70 % de la production de méthanal utilise ce procédé.
L'oxydation de l'éthanol avec le cuivre comme catalyseur conduit à l'éthanal.
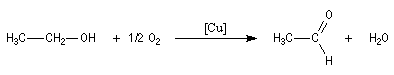
Oxydations avec coupure de la chaîne carbonée
Dans des conditions assez drastiques, les alcools secondaires cycliques sont
oxydés en cétones qui s'oxydent à leur tour avec rupture de la chaîne carbonée.
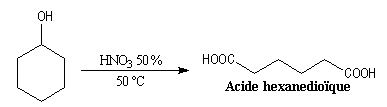
L'oxydation du cyclohexanol par l'acide
nitrique, permet la synthèse de l'acide hexane-1,6-dioïque encore appelé
acide adipique [3].
Oxydations utilisant le DMSO activé
Le DMSO
est largement utilisé en tant que solvant dipolaire aprotique dans de
nombreuses synthèses organiques. L'atome de soufre possède un certain caractère
électrophile comme le montrent les formes mésomères ci-dessous :
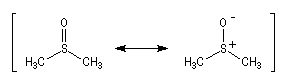
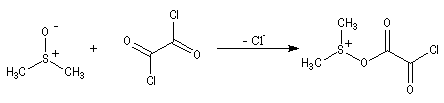
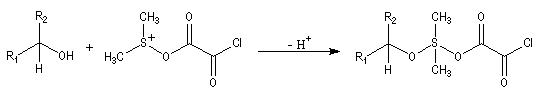
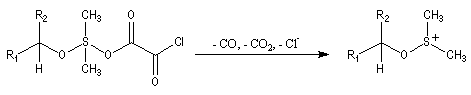
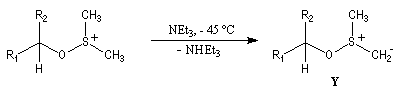
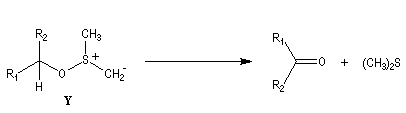
Oxydation d'Oppenauer
Il s'agit d'une méthode d'oxydation douce des alcools secondaires. On peut la
considérer comme la réaction inverse de la réduction de Meerwein-Pondorff
et Verley. Cette méthode a perdu une partie de son intérêt depuis la généralisation
de la méthode de Swern.
![]()
![]()
Propriétés acido-basiques
Propriétés acides
Les alcools ne manifestent aucune propriétés acides dans l'eau. Dans ce
solvant, l'ionisation de la liaison OH d'un alcool comme le méthanol est extrêmement
faible. Ainsi la constante thermodynamique de la réaction suivante vaut :
K = 10-16 à 25 °C.
![]()
Comme le produit ionique de l'eau est Ke = 10-14 à cette température, la constante thermodynamique de la réaction entre cet alcool et l'hydroxyde de sodium vaut K = 10-2.
![]()
L'ion hydroxyde ne peut donc produire l'ion méthanolate (et a fortiori les autres ions alcanolate) qu'en quantité catalytique. Inversement, les ions alcanolate sont des bases fortes nivelées.
|
|
|
Dans l'eau, les autres alcools sont moins acides que le méthanol. Les pKa des couples acido-basiques sont mesurés dans des solvants non-aqueux puis extrapolés en phase aqueuse. Les valeurs suivantes sont approchées :
|
Alcool |
CH3OH |
C2H5OH |
(CH3)2CHOH |
(CH3)3COH |
|
pKa (ROH/RO-) |
16 |
18 |
18 |
19 |
Plusieurs méthodes sont mises en œuvre pour déprotoner quantitativement les alcools.
|
Couple |
H2/H- |
NH3/NH2- |
|
pKa |
35 |
38 |
![]()
Avec les alcools primaires, les plus faciles à déprotoner, on utilise
le sodium :
E0 (Na+/Na) =
-2,7 V. Avec les alcools tertiaires comme le tertiobutanol qui sont moins réactifs
on utilise le potassium.
![]()
Avec les alcools primaires, les plus faciles à déprotoner, on utilise
le sodium :
E0 (Na+/Na) =
-2,7 V. Avec les alcools tertiaires comme le tertiobutanol qui sont moins réactifs
on utilise le potassium.
Propriétés basiques
L'atome d'oxygène des alcools manifeste des propriétés basiques. Les
alcools peuvent être protonés en présence d'un acide fort comme l'acide
sulfurique.
|
Alcool |
CH3OH |
CH3CH2OH |
(CH3)2COH |
|
pKa |
-2,2 |
-2,4 |
-3,8 |
Ces réactions sont surtout importantes lorsqu'elles précédent le départ d'eau en tant que nucléofuge.
Propriétés nucléophiles de l'oxygène
Synthèse de Williamson des éthers
L'atome d'oxygène des alcools pas suffisamment nucléophile pour déplacer
directement des nucléofuges moyens. Un moyen d'exalter la réactivité nucléophile
de l'oxygène est d'utiliser l'ion alcoolate. C'est la méthode utilisée dans la synthèse de A.W. Williamson des éthers.
La réaction qui suit un mécanisme SN2, est
accélérée dans les solvants dipolaires aprotiques comme le DMSO.
On peut, par cette méthode, synthétiser des éthers dissymétriques.
L'exemple ci-dessous concerne la préparation de l'éther méthylique du menthol.
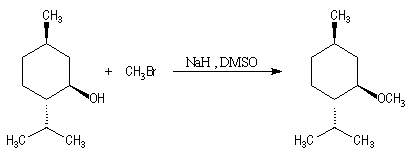
Cette méthode permet la synthèse des époxydes (oxacyclopropanes) en utilisant des halohydrines comme composés de départ. La réaction est une SN intramoléculaire.
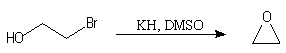
Les époxydes sont des composés importants car ils peuvent être ouverts par un grand nombre de réactifs nucléophiles (RMgX, RO-, LiAlH4, OH-, ...) Par une méthode analogue, on sait synthétiser des éthers cycliques à 3, 4, 5, 6, 7 chaînons. Les meilleurs rendements sont obtenus pour les cycles à 3, 5 et 6 chaînons.
La réaction de Williamson est également possible en utilisant OH- comme base, à condition de transférer cet ion en phase organique au moyen d'un agent de transfert de phase comme un ion ammonium quaternaire.Alcoolyse des halogénures tertiaires
La synthèse des éthers
dérivant de dérivés halogénés tertiaires est possible par un mécanisme SN1
s'il peut se former un carbocation relativement stable.
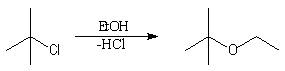
Hémiacétalisation, acétalisation
Synthèse des hémiacétals
La réaction entre un alcool et un aldéhyde ou une cétone conduit à un hémiacétals
(avec les cétones on utilise parfois le terme d'hémicétal mais cette
terminologie tend à disparaître). Le système est le siège d'un équilibre
qui est généralement en faveur du composé de carbonylé de départ. Cette réaction
est l'objet d'une catalyse acido-basique généralisée. On utilise souvent
l'acide paratoluène sulfonique (APTS) comme catalyseur.
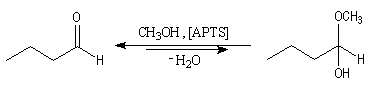
Synthèse des acétals
Les hémiacétals peuvent réagir avec un équivalent d'alcool pour
conduire, par une réaction également équilibrée, aux acétals.
Alors que la formation des hémiacétals est l'objet d'une catalyse acido-basique générale, celle des acétals est catalysée spécifiquement par les acides.
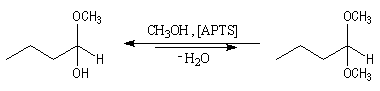
L'équilibre est défavorable au produit. Un moyen de déplacer sa position consiste à éliminer l'eau. On y parvient en ajoutant du toluène au mélange. L'eau et le toluène forment un hétéroazéotrope. On piège l'eau dans un décanteur.
Les acétals et les cétals sont des composés peu réactifs. Ils sont stables en milieu basique mais instables en milieu acide en présence d'un excès d'eau. Cette particularité permet de les utiliser comme groupes protecteurs des composés carbonylés ou des alcools.
Utilisation comme groupe protecteur
On utilise souvent un diol comme l'éthane-1,2-diol car avec ces composés, on
obtient des acétals cycliques. La réaction est alors thermodynamiquement moins
défavorable grâce à l'effet entropique (deux molécules conduisent à deux
molécules).
La séquence réactionnelle suivante illustre l'utilisation d'un groupe protecteur pour la fonction carbonyle dans une synthèse magnésienne d'alcool.
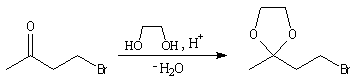
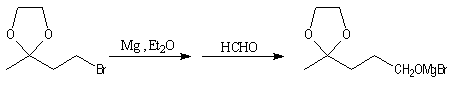
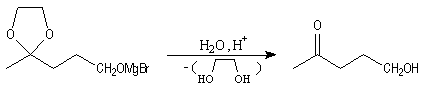
Acylation
Bilan
Une réaction d'acylation consiste formellement à substituer l’atome
d’hydrogène du groupe OH par un groupement acyle R-CO. On peut y
parvenir en effectuant la réaction entre l'alcool et l'acide carboxylique ou
l'un de ses dérivés : halogénure d'acyle, anhydride ou ester.
Avec les deux premiers, la réaction est à la fois totale et rapide. Avec l'acide ou l'ester elle conduit à un équilibre qu'on peut déplacer dans le sens de la formation de l'ester souhaité et nécessite l'utilisation d'un catalyseur.
Acylation par un chlorure d'acyle ou un anhydride
On effectue la réaction entre l'alcool et le chlorure d'acyle ou
l'anhydride en présence d'une base comme la pyridine, cela afin d'éviter de
former l'ester en milieu acide dans lequel il est instable.
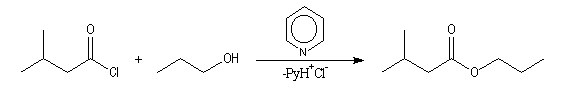
Le mécanisme de la réaction implique deux stades. Le premier stade est l'addition nucléophile de l'oxygène de l'alcool sur le carbonyle de l'halogénure d'acyle ou de l'anhydride.
L'intermédiaire tétraédrique formé se fragmente en éliminant l'ion halogénure, bon nucléofuge. Finalement l'oxygène est déprotoné par une base qui a été ajoutée dans le milieu réactionnel comme la pyridine, ou à défaut qui y est naturellement présente (Cl-). Avec un anhydride le résultat est analogue.
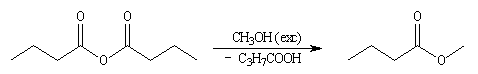
Acylation par un acide carboxylique
On appelle cette réaction l'estérification de Fischer. Son bilan s'écrit
:
La réaction est très lente à la température ordinaire, en l'absence de catalyse. On l'accélère en élevant T et en utilisant un acide fort tel que H2SO4comme catalyseur. Elle conduit à un état d'équilibre. Le pourcentage d'ester à l'équilibre dépend de la classe d'alcool :
La réaction étant quasiment athermique on ne peut espérer déplacer l'état d'équilibre en élevant la température. Pour favoriser la formation d'ester une solution est de travailler avec un excès de réactif ou bien d'éliminer l'un des produits au fur et à mesure de sa formation. Il y a deux possibilités :
|
|
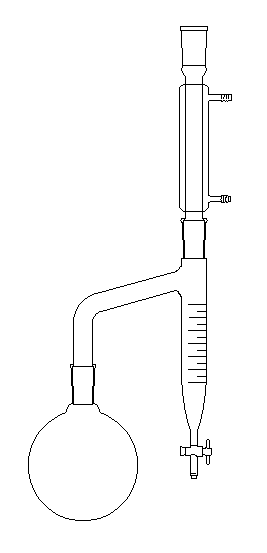
Le dessin de gauche représente un montage permettant la préparation d'un ester en utilisant un décanteur de type Dean-Stark. Dans le ballon, on réalise un mélange d'acide et d'alcool et de toluène (on utilise aussi le benzène mais l'usage de ce dernier répond à une réglementation précise du fait de sa toxicité). L'eau et le toluène ne sont pas miscibles à l'état liquide et ils forment un hétéroazéotrope. Le mélange de vapeur d'eau et de toluène monte dans le réfrigérant ascendant. Lorsque la température diminue, les vapeurs se liquéfient pour donner deux liquides non miscibles. L'eau, plus dense, tombe au fond du décanteur. Elle peut être extraite du milieu réactionnel au fur et à mesure de sa formation. Un mode opératoire possible pour la préparation de l'éthanoate de butyle est donné ci-dessous : On introduit dans le ballon 0,25 mol d'acide acétique (éthanoïque d = 1,05) et 0,25 mol de butan-1-ol (d = 0,81). On ajoute 30 mL de toluène et environ 0,15 g d'acide paratoluènesulfonique APTS ainsi que quelques grains de pierre ponce.Le mélange est chauffé avec un chauffe-ballon tant que l'eau est entraînée. Avec un appareil de Dean-Stark gradué on peut tracer la courbe donnant V en fonction du temps (ou de 1/t , c'est alors pratiquement une droite). |
Mécanisme de l'estérification de Fischer
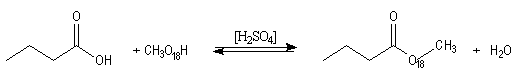
Le marquage isotopique (18O) de l'oxygène
de l'alcool, suivi de l'analyse par spectrométrie de masse des produits, montre
que cet atome se retrouve dans l'ester. Le mécanisme suivant s'applique aux
alcools primaires et secondaires. Il est détaillé ici dans le cas de la réaction
entre l'acide propanoïque et le méthanol.
Il s'agit d'un mécanisme d'addition-fragmentation. On distingue les différentes étapes suivantes qui sont renversables :
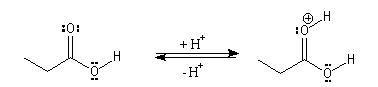
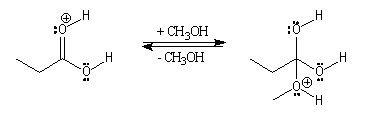
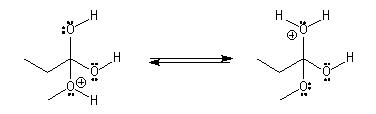
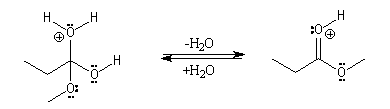
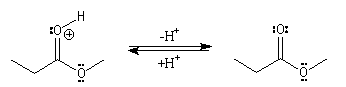
Notons que la concentration en H+ne doit pas être trop grande afin de ne pas protoner l’oxygène de l’alcool ce qui bloquerait son pouvoir nucléophile.
Estérification par l'acide nitrique
La nitroglycérine est le représentant le plus connu des nitrates d'alkyles. Ces composés (qui sont tous très instables et donc dangereux) sont obtenus par estérification des alcools par l'acide nitrique.
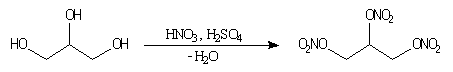
Comme la plupart des explosifs, un petit volume de nitroglycérine liquide libère un volume très élevé de gaz.

|
|
La nitroglycérine a été préparée pour la première fois par le chimiste italien A. Sobrero en 1846. Ce composé est particulièrement instable et peut exploser sous l'action d'une élévation de température ou d'un choc. Le suédois A. Nobel découvrit en 1866 qu'on pouvait stabiliser la nitroglycérine en la mélangeant à un sable siliceux d'origine naturelle (terre d'infusoire), le Kieselguhr. On l'utilise en médecine sous forme de patch comme vasodilatateur sous le nom plus rassurant de trinitrine. Les intérêts de la fortune accumulée par A. Nobel servent à distribuer les prix Nobel qui sont attribués depuis 1900 par l'Académie royale de Suède. |
La phosphorylation du glucose par l'ATP en glucose-6-phosphate est une importante réaction biochimique intervenant dans la glycolyse (coupure du glucose en pyruvate). Elle est catalysée par une enzyme spécifique, l'hexokinase
Coupure C-O
Formation de carbocations
Dans certains cas, la rupture de la liaison carbone-oxygène conduit à
former un carbocation relativement stable. Il est ainsi très facile d'obtenir
le carbocation triphénylméthyle à partir du triphénylméthanol par ajout
d'acide sulfurique.
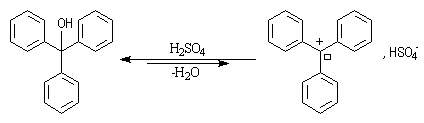
Dans le cas précédent on peut identifier le carbocation grâce à la couleur rouge que prend le milieu réactionnel.
|
|
A la température ordinaire, le triphénylméthanol est un solide de couleur blanche (TF = 136 °C). Dans le bécher j'ai mis une petite quantité de triphénylméthanol solide. L'ajout de quelques gouttes d'acide sulfurique concentré provoque l'apparition d'une couleur rouge intense. Cette expérience, réalisée en 1901 indépendamment par Norris et Kehrmann, fut la première mise en évidence de l'intervention de carbocations en chimie organique. L'acide sulfurique protone l'alcool et permet le départ du nucléofuge H2O. L'équilibre est déplacé vers la droite en raison de la stabilité du cation triphénylméthyle (carbocation trityle) et du caractère déshydratant de l'acide sulfurique concentré qui capte l'eau formée. |
Le cation triphénylméthyle forme un système conjugué de grandes dimensions. Ce système absorbe la lumière dans le domaine visible d'ou la couleur observée qui est approximativement complémentaire de celle qui est absorbée.
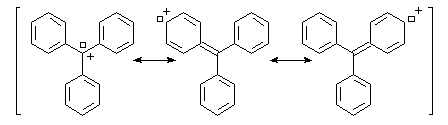
Halogénation par les hydracides halogénés
Alcools primaires
![]()
![]()
Alcools secondaires
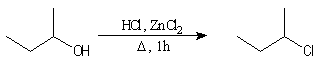
Alcools tertiaires
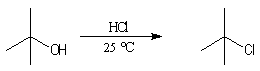
Test de Lucas des alcools : le rôle catalytique de ZnCl2 dans l'halogénation par les ions chlorure est à la base d'un test des classes d'alcools mis au point par le chimiste américain H. J. Lucas. Le réactif de Lucas est une solution de ZnCl2 dans l'acide chlorhydrique concentré. On effectue un mélange de l'alcool à tester et du réactif. Le test repose sur la différence de réactivité des alcools des différentes classes. Un test positif se traduit par l'apparition de deux phases car l'halogénure formé est peu miscible avec le réactif.
|
Classe de l'alcool |
Primaire |
Secondaire |
Tertiaire |
|
Vitesse |
très lent à chaud |
rapide à chaud |
rapide à froid |
Halogénation par des réactifs inorganiques
Réactifs halogénants
Les alcools peuvent être convertis en dérivés halogénés grâce à une
assez grande variété de réactifs halogénants: Un réactif couramment employé est le chlorure de thionyle SOCl2.
|
|
|
La réaction est souvent conduite en présence d'une amine tertiaire comme la pyridine pour capter HCl formé. Le dioxyde de soufre SO2 est un gaz dans les conditions de l'expérience. Il faut prévoir un piège à gaz acide. La méthode est utilisable avec les alcools primaires et secondaires.
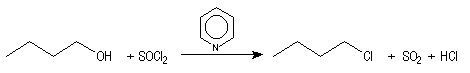
Les halogénures de phosphore sont des agents halogénants très efficaces.
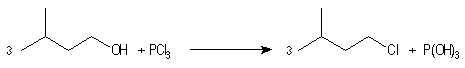
|
|
|
Le pentachlorure de phosphore PCl5 est parfois utilisé.
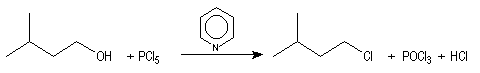
|
|
|
Cette méthode ne s'applique pas aux alcools possèdant des substituants en b. L'inconvénient est le faible pourcentage d'utilisation de l'élément chlore.
Bilan et conditions expérimentales
Une solution pour améliorer le caractère nucléofuge
du groupe OH est de le remplacer par un autre groupe ! Le chlorure de paratoluènesulfonyle
(TsCl) est un dérivé de l'acide paratoluènesulfonique (APTS).
On prépare le chlorure de paratoluènesulfonyle (chlorure de tosyle) grâce à la réaction suivante :
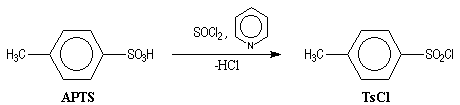
La réaction entre TsCl et un alcool conduit au paratoluène sulfonate ROTs (appelé souvent tosylate).
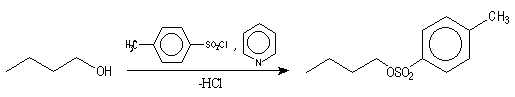
Ils peuvent être déplacés par de nombreux nucléophiles. L'ion CN- déplace facilement le groupe tosylate ce qui constitue une synthèse de nitrile.
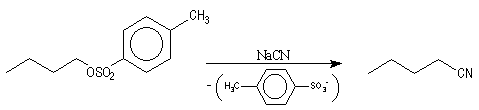
Notons que cette réaction ne serait pas possible en milieu acide dans lequel l'ion cyanure serait protoné. La méthode est surtout valable pour les alcools primaires et secondaires.
Elimination : passage aux composés éthyléniquesBilan, conditions expérimentales
Un moyen très simple pour préparer le cyclohexène consiste à chauffer le
cyclohexanol
avec de l'acide sulfurique ou de l'acide phosphorique concentrés. La réaction
s'écrit :
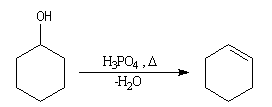
Cette réaction est générale. Les alcools conduisent aux composés éthyléniques par élimination d'eau. On peut regarder cette réaction comme l'inverse de l'hydratation de ces composés.
L'élimination peut être effectuée en présence d'un acide fort comme H2SO4 ou mieux H3PO4 . En milieu acide, l'alcool est protoné ce qui permet le départ d'eau bien meilleur nucléofuge que OH. On peut également utiliser des acides de Lewis tels que ZnCl2, BF3, I2 comme catalyseur de déshydratation.
Influence de la classe d'alcool
Des conditions typiques d'élimination à partir d'alcools de différentes
classes sont les suivantes :
|
Classe d’alcool |
Réactif |
Température (°C) |
|
Primaire |
H2SO4 (98 %) |
180 |
|
Secondaire |
H2SO4 (50 %) |
140 |
|
Tertiaire |
H2SO4 (20 %) |
80 |
La réaction est d'autant plus facile que la classe de l'alcool est plus élevée. Avec un alcool tertiaire un léger chauffage en présence d'acide dilué suffit à provoquer l'élimination.
Mécanismes
La déshydratation d'un alcool peut être regardée comme la réaction
inverse de l'hydratation
d'un alcène acido-catalysée. Avec les alcools tertiaires et secondaires,
il y a formation d'un carbocation. L'étape de formation de cet intermédiaire
est cinétiquement déterminante. La déprotonation du carbocation intervient
dans une seconde étape rapide.
Avec les alcools tertiaires, le mécanisme limite est E1. Avec les alcools primaires, le mécanisme limite est E2.
Régiosélectivité, stéréosélectivité
Les éliminations impliquant les alcools sont le plus souvent sous contrôle thermodynamique. On obtient majoritairement à l'équilibre le composé éthylénique le plus stable. Pour un composé éthylénique simple, c'est celui dont la double liaison porte le plus grand nombre de substituants donneurs. C'est la règle obtenue de façon empirique par le chimiste russe Zaytsev en 1875.
La stéréosélectivité s'explique par un contrôle thermodynamique de la réaction. La déshydratation du butan-2-ol fournit de façon majoritaire l'un des deux alcènes diastéréoisomères possibles. L'alcène de configuration E, plus stable que celui de configuration Z est obtenu majoritairement.
En revanche que le (2R)-butan-2-ol et le (2S)-butan-2-ol fournissent le même alcène. La réaction n'est pas stéréospécifique.
Le passage par des carbocations explique l'existence de réarrangements fréquents dans ce type de réaction.