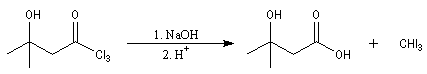|
|
|
Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
LES ALDÉHYDES ET LES CÉTONES
Généralités
Carbonylés importants|
|
|
|
|
|
|
|
La propanone est préparée à partir du propène par le procédé Wacker. |
Propriétés physiques
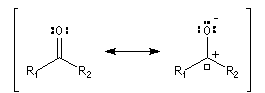
Structure électronique du groupe carbonyle
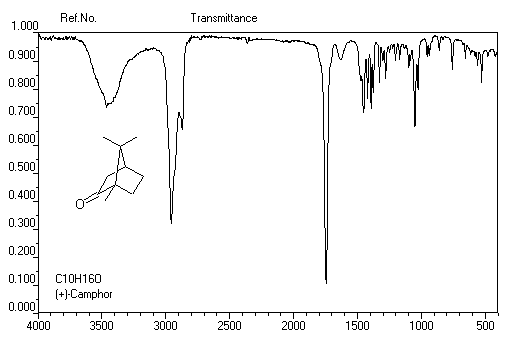
Spectroscopie infrarouge
C'est une méthode de choix pour l'étude des composés carbonylés. Le nombre
d'onde de la vibration d'élongation du groupe carbonyle dépend du type de
composé mais apparaît nettement sous la forme d'une bande intense dans une région
assez dégagée du spectre. Les aldéhydes possèdent en outre une absorption
caractéristique dûe à la vibration d'élongation de la liaison C - H et qui
se présente sous la forme d'un pic fin.
|
s (cm-1) |
2750 - 2900 |
1650 - 1730 |
|
Vibration |
Elongation C - H |
Elongation C = O |
On donne ci-dessous quelques exemples de spectres.

4-Méthylpentan-2-one
Benzophénone
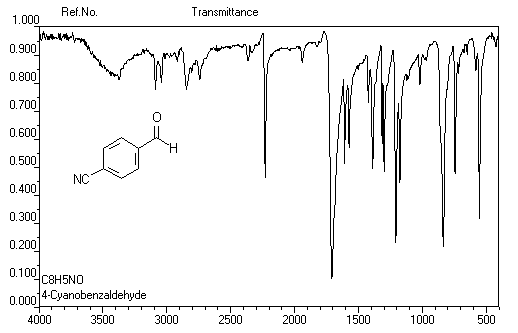
4-Cyanobenzaldéhyde
Notons une propriété importante concernant le nombre d'onde de la vibration d'élongation du carbonyle.
|
Protons |
H3CO- |
-CH2-CO- |
H-CO- |
|
d (ppm) |
2 |
2,5 |
10 |
On repère facilement les méthylcétones par la présence d'un pic vers 2 ppm. On notera le déblindage très important des protons aldéhydiques. L'effet inductif attracteur du groupe carbonyle diminue la densité électronique autour du noyau d'hydrogène.

Butan-2-one
Réactions d'additions ioniques sur le carbonyle
Généralités sur les additions ioniques
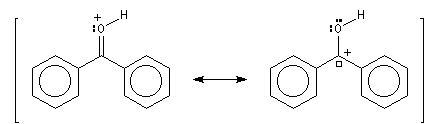
Lorsqu'on dissout de la benzophénone dans l'acide sulfurique concentré, la solution obtenue a une couleur rouge. On interprète cette apparition de la couleur par l'existence dans l'acide sulfurique d'une forme protonée qui accroit les possibilités de délocalisation électronique.
D'une façon générale en présence d'un acide de Lewis assez fort que nous noterons M+ on observe l'équilibre suivant :
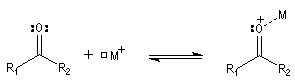

Deux cas peuvent se présenter :
Addition nucléophile des hydrures
![]()
LiAlH4 réduit de nombreuses doubles liaisons polarisées. Il est peu sélectif.
|
|
Le tétrahydruroborate de sodium NaBH4 est un agent réducteur beaucoup plus doux que LiAlH4. On peut le préparer en faisant réagir l'hydrure de sodium et le diborane.
|
NaBH4 est beaucoup moins réactif que LiAlH4. Il offre de ce fait, l'avantage d'une beaucoup plus grande sélectivité. On peut l'utiliser en milieu hydroalcoolique.
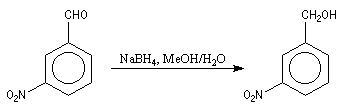
La réaction
de Meerwein, Ponndorf et Verley dans laquelle l'isopropylate d'aluminium
permet le transfert d'un ion hydrure vers un composé carbonylé. Les réactions
de réduction des carbonylés impliquant des organomagnésiens
encombrés procèdent d'un mécanisme assez comparable.
Stéréochimie en série cyclique
La réaction entre les hydrures métalliques et les cétones cycliques est diastéréosélective.
La stéréochimie de l'addition est gouvernée par des facteurs stéréo électroniques.
Avec les hydrures non encombrés comme NaBH4 ou
LiAlH4, le facteur électronique est prédominant
et la réaction normale correspond à une attaque axiale, conduisant à
l'alcool possédant un groupe OH équatorial. Avec les hydrures encombrés,
l'attaque équatoriale peut devenir prédominante. L'alcool possédant le groupe
OH axial est alors majoritaire.
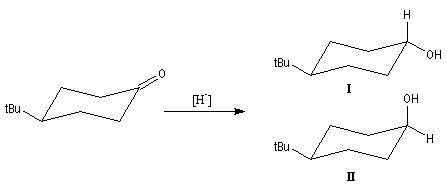
|
Composé |
I |
II |
|
LiAlH4 |
92 |
8 |
|
LiBH(secBu)3 |
7 |
93 |
Le cas des composés bicycliques est un plus délicat. Expérimentalement, on constate que les facteurs stériques sont prédominants et l'hydrure est transféré de façon préférentielle sur la face moins encombrée. L'exemple suivant concerne la réduction de la molécule de camphre qui donne un mélange dans lequel l'isobornéol (exo) est largement majoritaire sur le bornéol (endo).
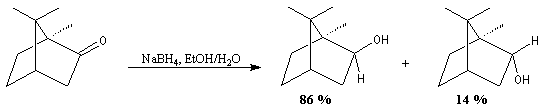
Induction asymétrique 1,2. Modèle de Felkin
Pour prévoir la stéréochimie de l'addition, plusieurs modèles de
l'état de transition ont été proposés. Le plus ancien est connu sous le nom
de règle
de Cram (1953).
Si l'on excepte l'hydrate de chloral qu'on peut obtenir à l'état cristallisé, la plupart des gem-diols sont des composés instables. D'un point de vue préparatif cette réaction offre donc peu d'intérêt. En revanche elle permet d'obtenir des informations intéressantes sur la réactivité comparée des composés carbonylés. Le tableau ci-dessous regroupe quelques valeurs pour la constante thermodynamique K de l'équilibre.
K = [hydrate]/[carbonylé]
|
Composé |
CH3COCH3 |
CH3CHO |
HCHO |
F3CCHO |
|
K |
1,4 10-3 |
1,06 |
2,3 103 |
2,9 104 |
On constate que vis à vis de l'hydratation, les aldéhydes réagissent plus facilement et plus complètement que les cétones. Cela peut s'expliquer par au moins deux facteurs :
La réaction d'hydratation est catalysée en milieu acide et en milieu basique.
En réalité, la situation est un peu plus complexe car on constate que les acides et les bases, même sous une forme non ionisée, catalysent cette réaction. On dit que la réaction est l'objet d'une catalyse acido-basique généralisée.
Oxydation des aldéhydes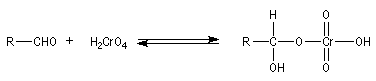
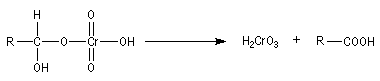
Addition des organométalliques
Classes d'alcool
Les réactions entre les composés carbonylés et les organométalliques
ont été traitées dans le chapitre correspondant. Rappelons simplement que ces
réactions constituent des méthodes de préparation des alcools appartenant aux
différentes classes.
|
Composé |
Méthanal |
Aldéhydes |
Cétones |
|
Classe d'alcool |
Primaire |
Secondaire |
Tertiaire |
Aspect stéréochimique
La stéréochimie de l'addition d'un organométallique sur les faces diastéréotopiques
d'un composé carbonylé peut être prévue en utilisant la règle
de Cram.
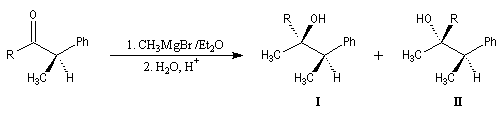
|
Ylure |
Non stabilisé |
Stabilisé |
|
Formule |
|
|
|
Préparation |
in situ |
à part |
EWG (electron widthdrawing group) désigne un groupe attracteur d'électrons.
La première étape de la préparation d'un ylure est la synthèse d'un sel de phosphonium. On réalise une subsitution nucléophile en utilisant un halogénure d'alkyle comme substrat. Le réactif le plus utilisé est la
triphénylphosphine. Cette réaction ressemble à l'alkylation d'Hofmann des amines. On notera cependant que le pouvoir nucléophile des amines est d'autant plus élevé qu'elles sont moins substituées alors qu'on observe l'inverse chez les phosphines. On peut attribuer cette différence à la taille plus importante de l'atome de phosphore par rapport à celle de l'atome d'azote.![]()
![]()
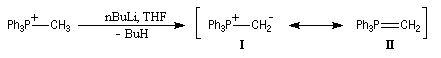
|
|
|
Mécanisme de la réaction de Wittig
Le mécanisme de la réaction de Wittig n'est pas connu dans tous ses détails
avec certitude. La réaction entre un ylure de phosphore et un composé carbonylé,
conduit à un intermédiaire cyclique appelé oxaphosphacyclobutane ou,
plus communément, oxaphosphétane.
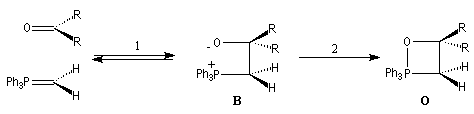
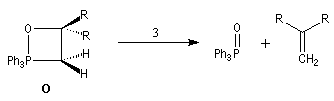
La prévision de la stéréochimie des produits de décomposition des
oxaphosphétanes constitue un problème délicat qui a fait l'objet de
nombreuses études. Plusieurs explications ont été avancées pour prévoir
cette stéréochimie. A l'heure actuelle ce problème n'est pas complètement résolu.
La décomposition des oxaphosphétanes est stéréospécifique.
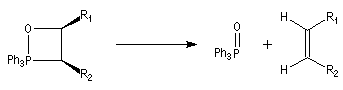
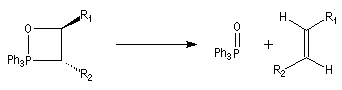
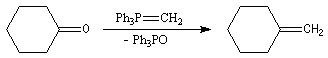
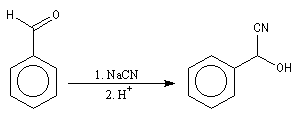
La réaction précédente peut être facilement renversée. Traités en milieu basique, les cyanhydrines redonnent le composé carbonylé parent.
Synthèse de Kiliani-Fischer Alcools
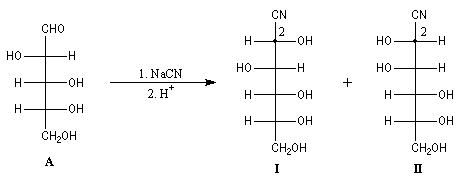
La formation de cyanhydrine peut servir à allonger la chaîne carbonée
d'un ose. Au cours de ses recherches visant à déterminer la structure du glucose,
le grand chimiste allemand E. Fischer l'a mise à profit pour préparer le
D-glucose à partir du D-arabinose.
L'addition de HCN au D-arabinose (A) fournit deux cyanhydrines diastéréoisomères
(I) et (II). Ces deux stéréoisomères diffèrent seulement par la
configuration de l'atome de carbone C2. Ce sont
des épimères.
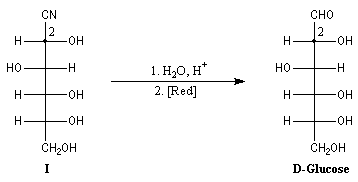
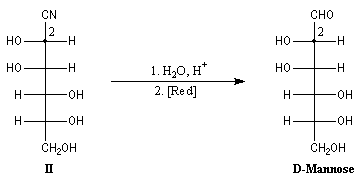
La réaction entre un alcool et un aldéhyde ou une cétone conduit à un hémiacétal.
La réaction qui est catalysée par un acide ou une base, aboutit à un équilibre
qui est généralement en faveur des réactifs.
Les hexoses comme le glucose,
existent principalement sous forme d'hémiacétals possédant 6 chaînons appelés
glucopyranoses.
La réaction entre un hémiacétal et un alcool, catalysée par un acide
fournit un acétal.
Du fait de leur faible réactivité, et du caractère renversable de leur réaction
de formation, les acétals sont utilisés comme groupe
protecteur des composés carbonylés ou des alcools. Les acétals sont
stables en milieu basique. En revanche ils redonnent facilement l'alcool et le
carbonylé parents par hydrolyse en milieu acide.
Thiols
Thiocétals
|
|
L'éthanedithiol est un réactif utilisé pour la synthèse des dithiocétals. On peut le préparer au moyen de la réaction :
|
Les thioacétals et les thiocétals peuvent être regardés comme les analogues soufrés des acétals et des cétals. Lorsque le but de la réaction est de préparer ces composés pour s'en servir d'intermédiaires, on peut traiter le composé carbonylé par l'éthanedithiol en présence d'un catalyseur du type acide de Lewis ZnCl2 .
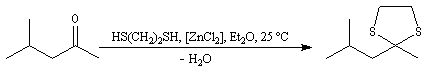
Les thioacétals et les thiocétals sont stables en milieu basique (une base très forte comme le nBuLi peut toutefois déprotoner les thioacétals). Ils sont auss
i raisonnablement stables en milieu acide.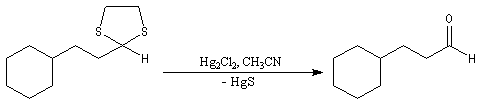
Amines
Imines
Dérivés azotés caractéristiques
Cas général
Nous allons symboliser le composé azoté par ZNH2.
Cette écriture permet de traiter simultanément plusieurs réactions qui procèdent
d'un mécanisme semblable
|
Z |
Réactif |
Produit |
Remarques |
|
H |
Ammoniac |
Imine non substituée |
Produit non isolable |
|
R |
Amine |
Imine substituée |
Produit isolable |
|
NH2 |
Hydrazine |
Hydrazone |
Produit solide |
|
NHPh |
Phénylhydrazine |
Phénylhydrazone |
Produit solide |
|
OH |
Hydroxylamine |
Oxime |
Produit solide |
|
NH-CO-NH2 |
Semi-carbazide |
Semi-carbazone |
Produit solide |
Le mécanisme de la réaction est détaillé ci-dessous. La vitesse de la réaction passe par un maximum pour un pH voisin de 4-5. Le rôle du milieu acide est de protoner le carbonyle de certaines molécules afin de les activer sans bloquer l'activité nucléophile du composé azoté.
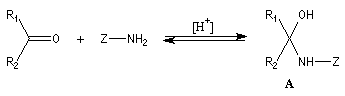

Lorsque Z est un groupe alkyle, le composé obtenu est une
imine substituée. Dans ce cas l'équilibre est plutôt en faveur des réactifs. La situation est différente quand la conjugaison entre la double liaison et un doublet non liant porté par Z permet une stabilisation du produit formé. C'est le cas pour les oximes, les hydrazones et leurs dérivés. Notons que ces composés présentent en général l'isomèrie Z, E.Oximes
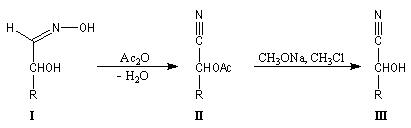
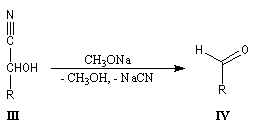
Les oximes sont obtenues en faisant réagir l'hydroxylamine avec un composé
carbonylé. Nous allons nous intéresser à la dégradation de Wohl qui permet
de diminuer d'une unité la longueur de la chaïne carbonée d'un ose.
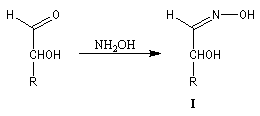
Hydrazones
L'hydrazine réagit avec les composés carbonylés pour donner des
hydrazones.
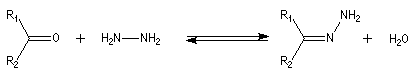
Réactions d'oxydo-réduction
Réduction en alcool
|
Composé carbonylé |
Méthanal |
Ethanal |
Cétones |
|
Classe d'alcool |
Primaire |
Secondaire |
Tertiaire |
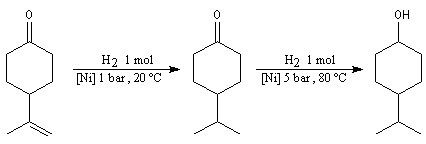
Hydrogénation catalytique
L'hydrogénation du groupe carbonyle nécessite des conditions plus dures
que celle d'une liaison éthylénique.
On peut ainsi réaliser des hydrogénations sélectives de doubles liaisons
éthyléniques en présence d'un groupe carbonyle.
Réduction de Meerwein-Ponndorf-Verley
Cette réaction assez ancienne procède d'un transfert
d'hydrure de l'isopropylate d'aluminium vers le dérivé carbonylé.
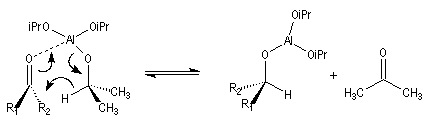
![]()
La réaction est renversable. Dans le sens inverse il s'agit d'une oxydation douce d'alcool connue sous le nom de
réaction d'Oppenauer.Réduction duplicative
Couplage pinacolique
Lorsque les conditions sont réunies pour que les
radicaux aient une durée de vie suffisante, on peut observer un couplage. Après
hydrolyse, on obtient un diol. Le magnésium est efficace dans ce genre de réaction
car on pense qu'il permet le rapprochement de deux molécules dans l'état de
transition.
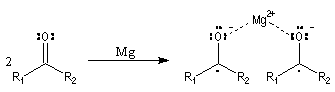

Réduction du carbonyle en méthylène
Réaction de Clemmensen Voici un exemple d'application de cette réduction.
La réaction consiste à traiter le composé carbonylé par un amalgame de
zinc en milieu acide, à chaud. Les électrons sont fournis par le métal.
L'acide joue le rôle de réactif protogène.
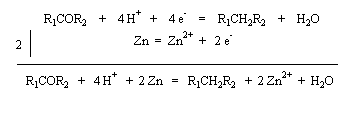
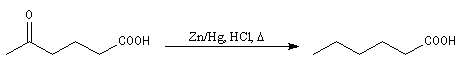
La réaction de Wolff-Kishner constitue une alternative intéressante à la
réaction précédente pour les molécules instables en milieu acide. Dans un
premier temps on commence par faire réagir le composé carbonylé avec
l'hydrazine. L'hydrazone
ainsi formée subit une réaction d'élimination de diazote par traitement
basique. Le bilan s'écrit :
![]()
Ce mode de réduction présente l'inconvénient de devoir travailler à une température élevée qui est peu compatible avec des structures fragiles. L'utilisation de tBuOK dont la basicité est exaltée par un solvant dipolaire aprotique comme le DMSO permet de réaliser la réaction d'élimination dans des conditions moins drastiques sur le plan thermique.
Le mécanisme de la réaction fait intervenir plusieurs stades :
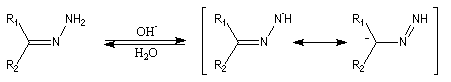
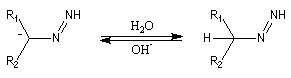
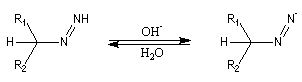

Oxydation
Aldéhydes
Les aldéhydes sont des composés très réducteurs. Ils
peuvent être oxydés par des oxydants doux. Les réactions suivantes utilisent
des complexes d'ions métalliques en milieu basique. Elles sont surtout utilisées
comme test analytique dans la chimie des sucres.
glucose.
![]()
![]()
Cétones
Les réactions d'oxydation des cétones cycliques
permettent une ouverture du cycle. Elle passent par l'intermédiaire de l'énol.
L'exemple suivant concerne l'ouverture de la cyclohexanone qui conduit à
l'acide hexane-1,6-dioïque.
Réactions au voisinage du carbonyle
Réalisées à partir du même composé carbonylé comme substrat, on constate expérimentalement que les réactions suivantes s'effectuent à la même vitesse.Si l'on traite la (S)-3-phénylbutan-2-one par une quantité catalytique d'acide ou de base, on obtient à l'équilibre un
mélange racémique de (S)-3-phénylbutan-2-one et de (R)-3-phénylbutan-2-one.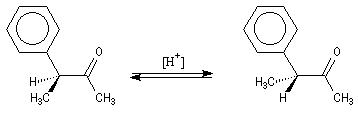
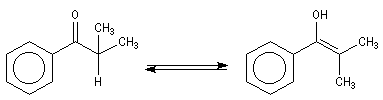
L'étape d'énolisation, commune aux réactions précédentes constitue l'étape cinétiquement déterminante ce qui explique les résultats expérimentaux précédents. L'énolisation est une manifestation de la mobilité de l'atome d'hydrogène porté par l'atome de carbone en a du groupe carbonyle. Le taux d'énolisation dépend du composé carbonylé et du solvant. Il est très faible pour les aldéhydes et les cétones simples mais la présence de l'énol à côté du composé carbonylé parent joue souvent un rôle essentiel dans de nombreuses réactions. Plusieurs méthodes parmi lesquelles la spectroscopie dans l'uv - visible et la RMN permettent de déterminer le pourcentage d'énol présent à l'équilibre. En milieu acide, le carbonyle est protoné. L'étape cinétiquement déterminante est la formation de l'énol.
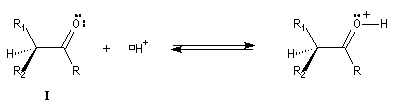
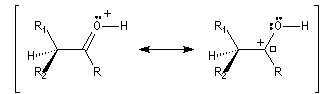
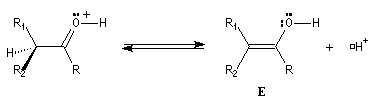
Taux d'énolisation
Le tableau ci-dessous regroupe quelques valeurs
expérimentales. Certaines particularités structurales peuvent expliquer un
pourcentage d'énol élevé.
On peut dégager deux tendances :

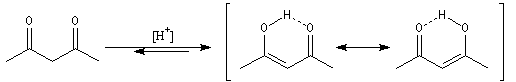
Acidité
Les composés carbonylés énolisables sont des
acides. L'atome d'hydrogène porté par l'atome de carbone en a
du carbonyle peut être arraché par une base suffisamment forte.
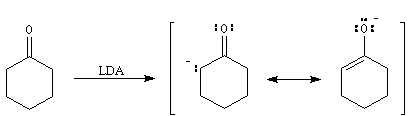
La propanone ne peut être déprotonée de façon quantitative que par des
bases très fortes en milieu non aqueux. Le butyllithium
ou le diisopropylamidure
de lithium (LDA) sont des bases couramment utilisées. En revanche, une dicétone
comme la pentane-2,4-dione est aisément déprotonée par la soude aqueuse. On
notera un accroissement très important (20 unités de pK) quand on passe du méthylpropène
à la propanone. Puisqu'il s'agit d'un équilibre, examinons l'état initial et
l'état final.
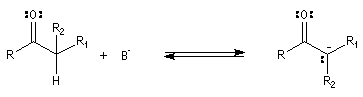
Alkylation des énolates
Alkylation des énolates
L'alkylation directe des aldéhydes
est peu pratiquée en raison de la réaction concurrente de condensation
aldolique. Examinons l'alkylation des cétones.
L'alkylation intramoléculaire est une méthode de formation de cycles qui
s'effectue avec un bon rendement.
Condensation aldolique
L'aldolisation est une réaction d'addition entre deux aldéhydes. Elle conduit à un équilibre. Le produit obtenu possède le nom évocateur d'aldol. La réaction inverse de coupure d'un aldol s'appelle rétroaldolisation. L'équilibre est en faveur du produit. Si l'on opère à température assez basse l'aldol peut être isolé.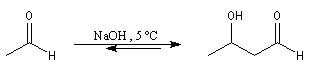
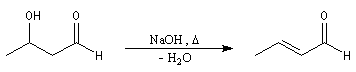
La déshydratation de l'aldol peut intervenir à la suite de l'aldolisation sans qu'on puisse isoler l'aldol intermédiaire. C'est le cas si la réaction intramoléculaire conduit à un cycle à 5 ou 6 chaînons.
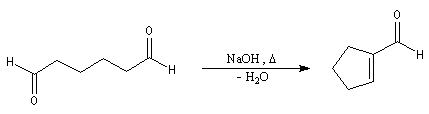
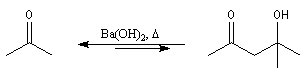

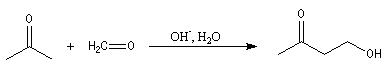
L'énolate obtenu réagit avec une cétone a-énone jouant le rôle d'accepteur de Michaël. On obtient un système dicarbonylé 1, 5.
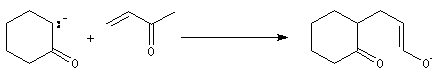
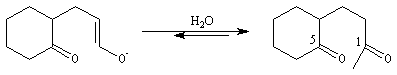
La dicétone obtenue subit une cyclisation par une réaction de cétolisation intramoléculaire.
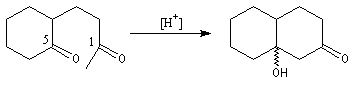
Le cétol obtenu est déshydraté ce qui fournit le produit final.
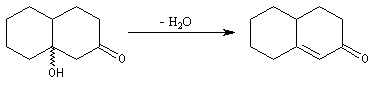
Halogénation
La réaction entre une cétone et le dibrome dans l'acide acétique conduit à une cétone a bromée. Dans ce milieu acide, la réaction peut être arrêtée au stade de la monobromation.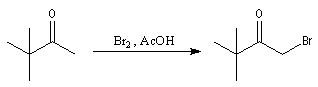
La réaction est régiosélective. L'halogénation a lieu préférentiellement sur l'atome de carbone le plus substitué.
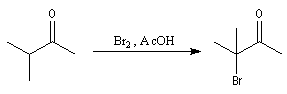
La réaction peut également avoir lieu en milieu basique mais ne peut généralement pas être arrêtée au stade de la monobromation.
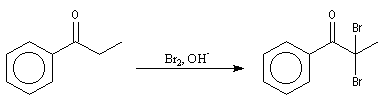

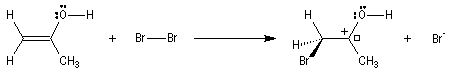
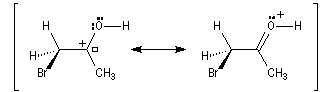
 c'est un test de présence
des cétones méthylées, c'est à dire ayant un des deux substituants au moins
qui est un -CH3. En récupérant du CHI3 jaune on sait qu'il y avait en présence
une cétone méthylée.
c'est un test de présence
des cétones méthylées, c'est à dire ayant un des deux substituants au moins
qui est un -CH3. En récupérant du CHI3 jaune on sait qu'il y avait en présence
une cétone méthylée.