|
|
|

Formule de Kékulé du benzène
En 1872, Kékulé perfectionna le modèle en suggérant que le benzène devait avoir une structure qui oscille entre deux formes. On passe d'une forme à l'autre en permutant les liaisons simples et doubles. Les tentatives répétées pour isoler ces formes et finalement leur échec ont ouvert la voie aux notions de conjugaison et de résonance.
La méthode de la résonance appliquée
au benzène
Énergie de résonance du benzène
L'hydrogénation du benzène s'effectue en bloc et conduit au cyclohexane.
Elle nécessite des conditions expérimentales assez fortes et un
catalyseur. Le Nickel Ni y est très efficace.

A température modérée, la réaction est totale.
Extension du concept d'aromaticité
[p]- annulènes
|
[p] |
4 |
6 |
8 |
|
nom |
cyclobutadiène |
benzène |
cyclooctatétraène |
- La molécule de cyclooctatétraène présente une infinité de conformations. La molécule adopte de façon préférentielle la conformation non plane (II).

Alors que le benzène est caractérisé par une grande stabilité (thermodynamique) et une inertie élevée (cinétique) en l'absence de catalyseur, le cyclooctatétraène possède une stabilité ordinaire et réagit beaucoup plus rapidement.
Pourquoi certains systèmes conjugués cycliques sont ils très stables tandis que d'autres ne le sont pas du tout ?
Méthode des orbitales moléculaires
Les niveaux d'énergie du cyclobutadiène (I), du benzène (II) et du cyclooctatétraène (III) sont donnés ci-dessous.
|
|
|
|
|
D E = 0 |
D E = 2 b |
D E = -1,65 b |



Effectuons le calcul dans le cas de la molécule de benzène. L'énergie EB du système p délocalisé est la somme des énergies des orbitales moléculaires occupées. Elle vaut :
DE = 2b
L'énergie de résonance électronique ER est par définition,
l'opposé de cette grandeur :
Critère d'aromaticité : règle de Hückel
Un hydrocarbure est aromatique s'il est : monocyclique, plan et qu'il possède 4n + 2 électrons délocalisables.
Le benzène est un hydrocarbure aromatique qui correspond à la valeur n = 1.
Un hydrocarbure est antiaromatique s'il est : monocyclique, plan et qu'il possède 4n électrons délocalisables.
Le cyclobutadiène (n = 1) et le cyclooctatétraène (n = 2) sont des hydrocarbures antiaromatiques.
Vérifications de la règle de Hückel
Après le benzène, le premier annulène qui satisfait la formule de Hückel
est le [10]- annulène :

Dans ce composé, il existe une répulsion importante entre les atomes d'hydrogène situés sur les atomes de carbone les plus proches, le [10]- annulène (I) a tendance à se cycliser en dihydronaphtalène (II) avec lequel il est en équilibre. Le composé n'est pas aromatique car il n'est pas plan.
L'hydrocarbure suivant qui satisfait la formule de Hückel est le [14]- annulène. Ce composé est moins tendu que le précédent mais il existe aussi sous deux formes en équilibre qui ne sont pas rigoureusement planes par suite des contraintes angulaires. Le [14]- annulène n'est pas, à proprement parler, aromatique.
L'hydrocarbure suivant est le [18]- annulène. Il possède cette fois un cycle suffisamment grand pour qu'une conformation plane puisse exister sans contrainte angulaire. Son spectre de RMN indique l'existence d'un courant de cycle diamagnétique qui montre son caractère aromatique.

Hydrocarbures polycycliques
Il existe de nombreux hydrocarbures possèdant des noyaux benzéniques accolés.
La parenté de ces composés avec le benzène entraîne pour certains d'entre-eux
un caractère aromatique indéniable (réactions de substitution électrophiles
etc). Cependant il faut faire attention que la règle de Hückel, qui n'est
valable que pour les hydrocarbures monocycliques, ne s'applique pas à ces
hydrocarbures .

|
composé |
naphtalène (I) |
phénantrène (II) |
coronène (III) |
|
électrons p |
10 |
14 |
24 |
Les deux premiers suivent la formule de Hückel mais pas le troisième malgré son caractère aromatique incontestable.
|
|
Le coronène possède 24 électrons p . La molécule est plane et possède un caractère aromatique incontestable. Le coronène ne satisfait pas la formule de Hückel. |
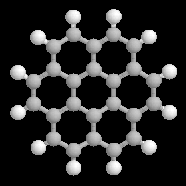
Hétérocycles aromatiques
Présentation
Un hétérocycle possède un ou plusieurs hétéroatomes (N, O, S etc) à
l'intérieur d'un cycle d'atomes de carbone. Le tableau suivant regroupe
quelques hétérocycles simples.
|
|
|
|
|
|
Pyridine |
Pyrrole |
Furanne |
Thiophène |




La pyridine et le pyrrole sont des amines hétérocycliques qui possèdent respectivement 6 et 5 chaînons. Ils sont tous les deux aromatiques avec 6 électrons délocalisés.
Dans la pyridine le doublet non liant de l'azote ne participe pas au système aromatique. En revanche dans le pyrrole ce doublet y participe.
|
|
C'est un composé toxique dont l'odeur est particulièrement écœurante. |

|
|
|

Ions aromatiques
Cation cyclopropéniumIl est également possible de préparer des systèmes conjugués cycliques cationiques. L'un des exemples les plus remarquables est le cation cyclopropénium qui bien que présentant des contraintes stériques importantes a pu être préparé. Il satisfait la règle de Hückel avec n = 0.

Le ferrocène
Par réaction entre l'ion cyclopentadiényle et les ions Fe2+
dans le DMSO on obtient un complexe dont la structure et les propriétés sont
remarquables : le ferrocène.

|
|
|

|
|
Le ferrocène est un composé organométallique. L'analyse par diffraction des rayons X et par diffraction des neutrons montre que la conformation la plus stable est celle-ci. |

Spectroscopie
Spectroscopie UV
Le spectre UV du benzène présente trois bandes d'absorptions :
- Bande E : lm = 185 nm, intense
- Bande E : lm = 204 nm, moyenne
- Bande B : lm = 260 nm, faible
La bande B possède une intensité faible car elle est interdite. On ne pourrait l'observer si la molécule était parfaitement rigide. Elle n'est visible que grâce au couplage avec les vibrations des liaisons entre les atomes (couplage vibronique).
Les spectres UV des polyacènes ressemblent à celui du benzène mais les longueurs d'onde des maxima d'absorption sont déplacés vers les grandes longueurs d'onde (effet bathochrome) et l'intensité des bandes est accrue.Spectroscopie de RMN
Le spectre ci-dessous est celui de l'isopropylbenzène (cumène). On distingue trois massifs qui correspondent à trois groupes de protons :
- d = 1,2 ppm doublet : 6 H des groupes méthyle
- d = 3 ppm septuplet : 1 H du groupe isopropyle
- d = 7,3 ppm multiplet complexe : protons du cycle aromatique

Spectroscopie infrarouge
Trois régions sont particulièrement intéressantes :
- 3030 cm-1 : vibrations d'élongation des H du phényle
- 1500 cm-1 - 2000 cm-1 : vibrations d'élongation du squelette aromatique
- 650 cm-1 - 1000 cm-1 : vibrations de déformation hors du plan des liaisons C-H
|
Composé |
Ortho |
Méta |
Para |
|
s (cm-1) |
730-770 |
750-810 |
800-860 |
Comme application, voici le spectre infrarouge de l'isomère ortho du 1,2-difluorobenzène.

Réactions d'addition sur les composés aromatiques
Complexes p
Les solutions de diiode dans le benzène sont violettes. Mais une étude fine du spectre d'absorption de la solution montre l'existence d'une bande intense dans le proche ultraviolet. Elle est due a la formation d'un complexe p dans lequel le benzène joue le rôle de donneur par son système d'électrons p et le diiode celui d'accepteur.
|
|
|

D'autres accepteurs forment des complexes de ce type avec les hydrocarbures aromatiques. Les autres halogènes : Br2 , Cl2. Le 2,4,6-trinitrophénol (acide picrique) forme des complexes cristallins avec certains aromatiques ce qui permet de les purifier.
Addition de dihydrogène
Application : hydrogénation sélective des chaînes
latérales
Le cycle est thermodynamiquement stable et cinétiquement beaucoup moins réactif
qu'une double liaison éthylénique. On peut donc hydrogéner sélectivement une
double liaison éthylénique en opérant à température et pression ambiantes.
L'exemple suivant concerne le styrène.

Addition du dichlore sur le benzène
L'addition du dichlore s'effectue en bloc. La réaction est initiée par un rayonnement UV, le mécanisme est radicalaire. On obtient différents stéréoisomères de l'hexachloro-1,2,3,4,5,6-cyclohexane. Puisque la molécule possède 6 atomes de carbone asymétriques, on peut prévoir un nombre maximum de 26 = 64 stéréoisomères. Compte-tenu de la symétrie de la molécule il y a seulement 9 stéréoisomères (7 composés meso et un couple d'énantiomères).

Le stéréoisomère ci dessous est appelé gammexan . C'est un puissant insecticide.
|
|
|
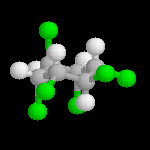
 L'image de gauche
représente le gammexan.
L'image de gauche
représente le gammexan.
Additions de Diels-Alder sur l'anthracène
Le cycle central de l'anthracène possède un caractère aromatique plus faible que les deux autres cycles. Il se comporte comme un diène et donne une réaction de Diels-Alder avec des diénophiles suffisamment réactifs comme le tétracyanoéthène TCNE.

|
|
|
|
|
I |
II |
III |



La réaction de Diels-Alder entre l'anthracène et le TCNE s'effectue par simple mélange des réactifs à la température ordinaire. L'erlenmeyer de gauche contient 0,3 g de TCNE (solution de couleur orange) dans 25 mL de toluène. L'erlenmeyer de droite contient 0,6 g d'anthracène dans 25 mL de toluène.
Ces composés sont tous toxiques. L'ensemble est placé sous une hotte ventilée. La réaction se manifeste pendant une période transitoire par une coloration verte due à un complexe de transfert de charge entre l'anthracène (donneur d'électrons) et le TCNE (accepteur d'électrons). Au bout de quinze minutes environ, on obtient le produit qui se présente sous la forme d'un composé solide à la température ordinaire (sublimation à 269 °C).Oxydo-réduction des cycles aromatiques
Oxydation du Benzène
L'oxydation du benzène est une réaction d'intérêt essentiellement industriel. On obtient l'anhydride maléique.

Oxydation de l'anthracène
Elle conduit à la formation d'une quinone, l'anthraquinone, ce qui témoigne du faible caractère aromatique du cycle central de cette molécule.

Réduction de Birch
Résultats expérimentaux
La réaction de Birch est une réduction des composés aromatiques par un métal
alcalin dissous dans l'ammoniac liquide ou une amine. Il s'agit d'une méthode
d'hydrogénation partielle, régiosélective, d'un cycle aromatique.
|
|
|

Mécanisme
Dans le cas du naphtalène, le mécanisme suivant peut être proposé :
- l'un des cycles accepte un électron ce qui conduit à la formation d'un anion-radical responsable de la couleur verte ;
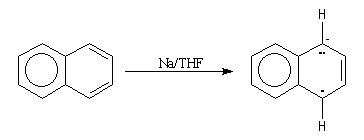
- le radical-anion réagit avec l'éthanol, solvant protogène pour conduire à un radical ;

- le radical précédent accepte un nouvel électron pour former un anion ;

- l'anion réagit à son tour avec le solvant pour donner le produit final.

Réactions de la chaîne latérale
Oxydation
Réaction
La réaction affecte la chaîne au niveau de l'atome de carbone en a
du cycle. Cette position benzilique est très sensible à l'oxydation dans le
cas où il y a au moins un atome d'hydrogène. La coupure de la chaine
carbonée s'effectue de manière régiosélective quelle que soit sa longueur.
L'acide obtenu est donc l'acide benzoïque.

Tout oxydant assez puissant peut convenir : KMnO4, K2Cr2O7, ou l'oxygène de l'air avec un catalyseur adéquat contenant du Co(II)
Applications
C'est une voie d'accès commode car l'introduction directe du groupe -COOH dans un cycle n'est pas possible par
substitution. L'oxydation du 1,4-diméthylbenzène (p-xylène) est une préparation
industrielle de l'acide 1,4-benzènedicarboxylique (acide téréphtalique).

Halogénation
Lorsqu'on porte à reflux du toluène et du dibrome à la température ambiante
sous irradiation UV, on obtient facilement le 1-bromo-1-phénylméthane.

Avec une chaîne latérale plus longue, la réaction est régiosélective et l'halogénation implique exclusivement la position benzylique. Cette régiosélectivité s'explique par l'intervention de radicaux benzyliques dans l'étape déterminant la vitesse. La chloration de la chaîne latérale est également possible mais elle est beaucoup moins régiosélective.
Réactions de substitutions électrophiles
Le substrat est le composé aromatique. Le réactif est un électrophile. La réaction consiste en la substitution d'un atome d'hydrogène du substrat par l'électrophile. Il s'agit donc d'une substitution électrophile en série aromatique. Elle est notée SEAr. Puisqu'il s'agit d'une substitution, le caractère aromatique du substrat se retrouve dans le produit final.Résultats expérimentaux
- La chloration et la bromation peuvent être effectuées par union directe entre le benzène ou l'hydrocarbure aromatique et les corps simples Cl2 ou Br2 à condition d'utiliser un acide de Lewis : AlCl3, AlBr3, FeBr3 comme catalyseur.
- La fluoration directe est impossible car F2 détruit le substrat par oxydation.
- L'iodation directe est également impossible car I2 n'est pas suffisamment réactif. L'iodation est réalisable en utilisant des réactifs qui sont source d'iode cationique comme le chlorure d'iode ICl (réactif de Wijs) ou le mélange I2/ HNO3 (car dans ce cas I- est oxydé par l'acide nitrique). Les dérivés iodés sont obtenus de manière plus commode en utilisant les sels de diazonium comme intermédiaires.
|
|
AlCl3 est un composé de couleur blanche, solide à la température ordinaire. Il est très hygroscopique et en présence d'eau ou d'air humide, il est hydrolysé en Al(OH)3 et HCl qui forme un brouillard.
|

Mécanisme
Nous allons raisonner sur l'exemple de la bromation. La réaction s'effectue
en plusieurs stades :
- Formation de l'électrophile : elle met en jeu la polarisabilité de la molécule de Br2. L'interaction avec l'acide de Lewis induit une polarisation de la liaison Br-Br.

- Formation d'un intermédiaire chargé de haute énergie appelé complexe de Wheland ou complexe s. Cette étape est l'étape cinétiquement déterminante de la réaction.

Ce complexe s possède une énergie beaucoup plus élevée que les réactifs. On peut décrire en première approximation sa structure électronique en utilisant la méthode de la mésomérie.

- Restauration de l'aromaticité.
Dans cette étape Br- joue le rôle de base.
La réaction de nitration du benzène peut être effectuée à la température ambiante en utilisant HNO3 fumant ou le mélange HNO3, H2SO4 concentré. Elle conduit au mononitrobenzène. Ce dernier, plus dense que les réactifs, va au fond du réacteur. Avec des aromatiques plus réactifs comme les phénols, l'acide nitrique dilué suffit mais le mécanisme est différent.
Mécanisme
Il s'agit d'une réaction par stades :
- Formation de l'électrophile : l'acide sulfurique joue un double rôle. C'est un réactif protogène qui améliore le caractère nucléofuge du groupement OH de l'acide nitrique en le protonant.


Le véritable réactif électrophile est le cation nitronium NO2+. On dispose de plusieurs preuves de l'existence de cet ion comme agent nitrant.
- Deuxième stade : formation de l'intermédiaire de Wheland.

- Troisième stade : restauration de l'aromaticité.
B- symbolise une base du milieu réactionnel HSO4- ou NO3-.
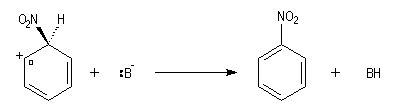
|
|
L'image de gauche représente le cation NO2+. Conformément aux prévisions de la méthode VSEPR, cet ion est linéaire.
HNO3 + 2 H2SO4 = NO2+ + H3O+ + 2 HSO4-
|

Les dérivés nitrés en tant qu'explosifs
Les dérivés polynitrés, notamment le 2,4,6-trinitrotoluène (TNT), sont utilisés
comme explosifs. On les obtient dans des conditions plus dures que les mononitrés.
La réaction doit être effectuée à chaud et sous pression.
|
|
|

Réduction des dérivés nitrés
Les dérivés nitrés aromatiques sont une voie d'accès intéressante aux
amines aromatiques par réduction du groupe nitro. L'introduction directe d'un
groupe amino n'est en effet possible que sur des cycles fortement appauvris en
électrons ou en utilisant des bases très fortes, en suivant un mécanisme de substitution
nucléophile aromatique SNAr.
La réduction du nitrobenzène par un métal en milieu acide, conduit à un sel d'anilinium. Après passage en milieu basique, on obtient l'aniline.

Au laboratoire on utilise l'étain Sn ou l'amalgame Zn / Hg en milieu chlorhydrique. Dans l'industrie on utilise le Fe comme réducteur du fait de son coût moins élevé.
Résultats expérimentaux
La sulfonation du benzène peut être effectuée en portant à reflux un mélange
de benzène et d'oléum
(SO3 dissous dans H2SO4 concentré).
Mécanisme
Il s'agit d'une réaction par stades. On va se placer dans le cadre d'un
contrôle cinétique de la réaction.
- Formation de l'électrophile : l'électrophile est le trioxyde de soufre SO3 ou sa forme protonée HSO3+. Nous allons raisonner avec SO3.

- Formation de l'intermédiaire de Wheland.

- Réaromatisation.

|
|
|

Défonctionnalisation des dérivés sulfonés
La réaction de sulfonation présente l'importante caractéristique
de pouvoir être renversée. Si l'on traite un acide sulfonique à chaud par une
solution étendue d'acide, on observe le remplacement de HSO3+
par H+. La force motrice de la réaction est, dans ce cas, la
formation des composés les plus stables. La réaction est thermodynamiquement
contrôlée.
Sulfonation du naphtalène
Par monosulfonation du naphtalène, on peut former a priori deux isomères
: l'acide naphtalène-1-sulfonique et l'acide naphtalène-2-sulfonique nommés
respectivement a et b.
Lorsque la réaction est effectuée à 50 °C on obtient de façon majoritaire le composé le plus rapidement formé : c'est l'isomère a qui est le produit cinétique de la réaction.

Si la réaction est effectuée à une température plus élevée 180 °C et se prolonge suffisamment pour qu'un équilibre s'établisse on obtient majoritairement l'isomère b. C'est le produit thermodynamique de la réaction.

Application de la réaction de sulfonation et des dérivés sulfonés
En raison du caractère renversable de la réaction, un acide sulfonique
peut être désulfoné par hydrolyse à température élevée. Cette caractéristique
permet d'utiliser la suite sulfonation-désulfonation pour bloquer
temporairement une position d'un cycle aromatique dans une synthèse multiétapes.
Les acides sulfoniques sont des acides forts qui possèdent de nombreuses
applications. L'acide paratoluènesulfonique, en abrégé
Alkylation de Friedel et Craft
Résultats expérimentaux
Il s'agit d'une réaction de création de liaison carbone-carbone. Le réactif
alkylant possède un atome de carbone déficient en électrons donc électrophile.
La réaction nécessite la plupart du temps l'emploi d'un acide de Lewis
comme catalyseur afin d'exalter le caractère électrophile du réactif. On
utilise AlCl3 ou AlBr3 avec les dérivés halogénés. H+
avec les alcools et les éthyléniques.
La réaction entre le benzène et le tétrachlorométhane en présence de AlCl3 constitue un moyen de synthèse du chlorure de triphénylméthyle.

|
|
Réaction de Friedel et Crafts entre le toluène et CCl4. |

Mécanisme
Il s'agit d'une réaction par stades mais les différentes étapes sont renversables.
On va raisonner avec un dérivé halogéné.
- Formation de l'électrophile :
La réaction entre le dérivé halogéné et un acide de Lewis (AlCl3 , AlBr3) conduit à un carbocation potentiel (ou à un vrai carbocation dans le cas où ce dernier est suffisamment stable).

- Formation de l'intermédiaire de Wheland.

- Réaromatisation.

![]()
Utilisation de la réaction de Friedel et Craft
La réaction est d'un emploi assez limité en synthèse car il y a souvent
ambiguité sur la nature du produit final. Les causes principales sont :
- la tendance marquée des carbocations aux réarrangements : primaire ® secondaire ® tertiaire ;
- il est très difficile d'éviter la polyalkylation car les dérivés alkylés réagissent plus rapidement que le benzène lui même ;
- les différentes étapes du mécanisme étant renversables, la réaction est la plupart du temps thermodynamiquement contrôlée ; le composé le plus stable est alors majoritaire.
Résultats expérimentaux
Il s'agit d'une synthèse de cétone aromatique. Le réactif acylant est un halogénure
d'acyle ou un anhydride d'acide. La réaction nécessite
l'utilisation d'un acide de Lewis comme AlCl3 en tant que catalyseur.

Mécanisme
On va raisonner dans le cas où le réactif acylant est un halogénure
d'acyle. On distingue trois étapes :
- Formation de l'électrophile : le caractère électrophile du carbone du chlorure d'acyle est exalté par la formation d'un complexe avec le catalyseur.

On peut aussi observer l'intervention d'un véritable cation acylium.

- Formation de l'intermédiaire de Wheland

- Restauration de l'aromaticité.

Formation d'un complexe avec le catalyseur
Quand on effectue l'acylation en présence d'un catalyseur comme AlCl3,
il est nécessaire d'utiliser au moins une mol de catalyseur par mole de cétone
formée. En effet celle-ci est suffisamment basique pour former un complexe avec
AlCl3 qui bloque son activité catalytique.

La cétone est libérée du complexe par traitement en milieu acide.

Applications
L'acylation constitue une voie d'accès aux cétones aromatiques mais elle
constitue aussi une alternative intéressante à la réaction d'alkylation de
Friedel et Craft. La réduction du groupe carbonyle en méthylène étant réalisable
assez facilement la plupart du temps. Il n'y a pas d'ambiguité sur le produit
final car les ions acylium ne se réarrangent pas.
Réaction d'acylation intramoléculaire
Dans les cas favorables (cycles à 5 ou 6 chaînons), les réactions
d'acylation peuvent être mises à profit pour réaliser des cyclisations, comme
le montre l'exemple ci-dessous :

Résultats expérimentaux
On peut se demander s'il est possible de synthétiser des aldéhydes en
utilisant la réaction de Friedel et Crafts. La formylation des hydrocarbures aromatiques est
possible en utilisant un mélange de monoxyde de carbone CO et de
chlorure d'hydrogène HCl avec AlCl3 comme catalyseur. La réaction prend
alors le nom de réaction de Gattermann-Koch. Avec AlCl3 seul, la réaction
est effectuée sous pression et elle est surtout utilisée dans l'industrie.

Le groupe phényle oriente la substitution sur les positions ortho et para de l'autre cycle. L'obtention quasi-exclusive de l'isomère para est dûe à l'encombrement de la position ortho et sera justifiée plus loin.
Substitutions électrophiles sur des dérivés du benzène
Position du problème
On s'intéresse à la réaction de SEAr entre un dérivé
monosubstitué du benzène Ph-A et un réactif électrophile. Ce dernier sera
noté symboliquement E+ mais il peut s'agir d'une molécule. Nous
allons aborder ce problème :
régiosélectivité : 3 stéréoisomères peuvent être obtenus dans des proportions qui sont a priori différentes.
Les positions relatives des substituants dans les dérivés disubstitués sont données par des noms consacrés par l'usage :|
Position relative |
1, 2 |
1, 3 |
1, 4 |
|
Nom |
Ortho |
Méta |
Para |
Examinons les résultats expérimentaux relatifs à la nitration de quelques composés aromatiques. Les résultats sont regroupés dans le tableau suivant :
|
Groupe |
Vitesse relative |
Ortho |
Méta |
Para |
|
-OH |
103 |
40 |
<2 |
58 |
|
-CH3 |
25 |
58 |
4 |
38 |
|
tBu |
16 |
12 |
8 |
80 |
|
H |
1 |
- |
- |
- |
|
-CH2Cl |
0,71 |
32 |
15,5 |
52,5 |
|
-Cl |
3,3 10-2 |
31 |
<0,2 |
69 |
|
-COOEt |
3,7 10-3 |
2 |
72 |
4 |
|
-CF3 |
2,6 10-5 |
6 |
91 |
3 |
|
-NO2 |
6 10-8 |
5 |
93 |
2 |
|
-N+(CH3)3 |
1,2 10-8 |
0 |
100 |
0 |
Les résultats précédents montrent qu'on peut classer les groupes A en deux catégories :
- ceux pour lesquels la vitesse relative est augmentée par rapport au benzène : les groupes sont qualifiés d'activants
- ceux pour lesquels la vitesse relative est diminuée par rapport au benzène : ils sont désactivants.
| Activants forts |
Activants faibles |
Désactivants faibles |
Désactivants forts |
|
-NH2 , -NHR , -NR2 , -NHCOR, -OH, -OR |
alkyle , phényle
|
-F, -Cl , -Br , -I |
-NO2 , -CF3 , -N+R3 -COOH, -COOR, -COR -SO3H, -CN |
L'examen de la régiosélectivité de la réaction permet également de distinguer deux catégories
- les groupes qui orientent en ortho ou para ;
- les groupes qui orientent en méta.
|
Orientation en ortho-para |
Orientation en méta |
|
activants forts activants faibles halogènes |
désactivants forts |
On va rationaliser ces résultats expérimentaux dans le cadre du modèle ci-dessous :
Modèle utilisé
Nous allons raisonner dans le cas fréquent où la réaction est sous
contrôle cinétique et dans laquelle la formation du complexe s
(intermédiaire de Wheland) constitue l'étape cinétiquement déterminante.
Dans ce cadre, le postulat de Hammond permet de ramener cette discussion à celle de la stabilité de l'intermédiaire de Wheland. Ce modèle n'est pas toujours satisfaisant dans les cas suivants :
- réactions de sulfonation et d'alkylation de Friedel et Craft qui sont fréquemment thermodynamiquement contrôlées ;
- réactions pour lesquelles l'étape cinétiquement déterminante n'est pas la formation de l'intermédiaire.
Nous allons limiter l'étude à celle des trois cas classiques de substitution sur les positions ortho, méta, para.
Stabilité de l'intermédiaire de Wheland
Dans le cadre du modèle prédent, l'intermédiaire de Wheland se forme dans l'étape
cinétiquement déterminante de la réaction. Nous allons comparer la stabilité
des différents intermédiaires possibles en utilisant comme outil la méthode
de la mésomérie.



Classement des groupes selon leur effet électronique
On peut distinguer quatre catégories :
|
Effet électronique |
Cinétique de réaction |
Régiosélectivité |
|
-I & -I, -M |
désactivants |
orientation méta |
- La réaction est ralentie par rapport à ce qu'on observe avec le benzène.
- L'orientation méta est la moins défavorable.
|
Effet électronique |
Cinétique de réaction |
Régiosélectivité |
|
-I, +M (sauf halogènes) |
activants |
orientation ortho-para |
Ce cas est plus délicat car les effets sont opposés. Si l'on met à part les halogènes qui seront étudiés plus loin. On peut retenir les résultats suivants :
- la vitesse de la réaction est plus grande qu'avec le benzène ;
- l'orientation ortho-para est favorisée.
|
Effet électronique |
Cinétique de réaction |
Régiosélectivité |
|
halogènes (-I, +M) |
désactivants |
ortho-para |
La réaction est ralentie par rapport au benzène (assez peu). L'orientation ortho-para reste favorisée.
|
Effet électronique |
Cinétique de réaction |
Régiosélectivité |
|
alkyles (+ I) |
activants |
ortho-para |
La réaction est accélérée par rapport au benzène. L'orientation ortho-para est favorisée.
Le rapport ortho-para
Le rapport r(o)/r(p) dépend beaucoup des conditions expérimentales : par
exemple dans la chloration du toluène, ce rapport varie selon les protocoles
expérimentaux, de 62/38 à 34/66. Plusieurs facteurs entrent en jeu :
- Le premier est de nature statistique.
Il existe en effet deux positions ortho pour une position para. Sur une base purement statistique on devrait s'attendre à 67 % pour l'isomère ortho et à 34 % pour le para. - Le second est de nature stérique.
On donne ci-dessous les rapports r(o)/r(p) pour la nitration d'alkylbenzènes avec des substrats possèdant des chaînes alkyles de plus en plus volumineuses :
|
Groupe |
Ortho |
Para |
r (ortho) / r (para) |
|
-CH3 |
58 |
37 |
0,78 |
|
-CH2CH3 |
45 |
49 |
0,46 |
|
-CH(CH3)2 |
30 |
62 |
0,24 |
|
-C(CH3)3 |
16 |
73 |
0,11 |
Pour un même substrat, ici le tertiobutylbenzène, le rapport suit le même type d'évolution lorsque la taille du groupe entrant augmente.
|
Réaction |
Ortho |
Para |
r (ortho) / r (para) |
|
chloration |
39 |
55 |
0,35 |
|
nitration |
30 |
70 |
0,21 |
|
bromation |
11 |
87 |
0,06 |
|
sulfonation |
< 1 |
> 99 |
0,005 |
- L'isomère ortho peut être favorisé quand une interaction stabilisante intervient entre les deux groupes. Par exemple une liaison hydrogène intramoléculaire, comme dans l'aldéhyde salicylique.
- Quand le groupe orienteur possède un doublet non liant, des effets plus fins peuvent augmenter le pourcentage de para au dépens de l'ortho. Les intermédiaires de Wheland possèdant une structure quinonique para sont en effet plus stables que ceux qui possèdent une structure quinonique ortho. Ce fait est confirmé en RMN qui donne des renseignements sur la distribution des charges sur les différents sommets du cycle.
Réactions de substitutions nucléophiles aromatiques
Mécanisme d'addition-élimination
Résultats expérimentaux
Les réactions de substitution nucléophiles sur des substrats aromatiques
sont possibles sur des cycles fortement désactivés par des groupes attracteurs
situés en ortho ou para du nucléofuge.


Mécanisme
Raisonnons sur le 1-chloro-2-nitrobenzène en tant que substrat.
- Addition du nucléophile et formation d'un complexe. Cette étape est l'étape cinétiquement déterminante
de la réaction. La relative stabilité de cet intermédiaire est due à la
présence du groupe nitro attracteur (-I, -M).

- Élimination du nucléofuge et formation du produit à partir du complexe.

- Synthèse de la 2,4-DNPH
La 2,4-DNPH est un réactif bien connu des composés carbonylés avec lesquels elle forme des 2,4-dinitrophénylhydrazones qui sont généralement facilement cristallisables et dont on peut déterminer le point de fusion.
- Identification des amino-acides N-terminaux des protéines
Le dinitrofluorobenzène (ou réactif de Sanger) peut être utilisé pour déterminer la nature de l'amino-acide N-terminal d'une protéine. Le groupe amino est suffisamment nucléophile pour déplacer le fluorure du DNFB par une réaction de SNAr.
Après hydrolyse, les fragments sont isolés. L'amino-acide N-terminal est identifié car c'est le seul qui a formé une liaison avec le réactif (les amides ne réagissent pas).
En opérant de la sorte, de proche en proche, on peut remonter à la structure primaire de la molécule. C'est en utilisant une méthode de ce type que le chimiste Britannique F. Sanger (Université de Cambridge) parvint en 1955 à élucider la structure primaire de l'insuline. Ses travaux lui valurent le prix Nobel de chimie en 1958.
Mécanisme d'élimination addition
Résultats expérimentaux
Lorsqu'on fait réagir le parabromotoluène par l'ion amidure dans
l'ammoniac liquide on obtient un mélange de deux composés : la paratoluidine
et la métatoluidine. La formation de la paratoluidine peut s'expliquer par la
substitution d'un atome de brome par un groupe amino. Celle de la métatoluidine
est plus surprenante car le groupe amino s'est fixé sur une position non
occupée du cycle aromatique.

Mécanisme
Le mécanisme de la réaction est différent du précédent car il n'existe
pas ici de groupe attracteur sur le cycle apte à stabiliser une charge négative.
Pour expliquer la formation des produits obtenus, J.D. Roberts a
imaginé dans les années 70, l'existence d'un intermédiaire possèdant deux
points d'attaque : le benzyne.
- formation de l'intermédiaire ;

- réaction entre le benzyne et le nucléophile ;

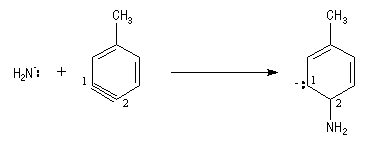
- restauration de l'aromaticité ;

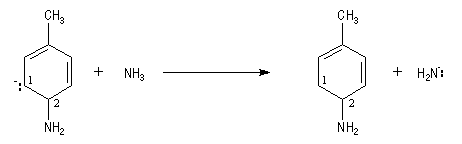
|
|
|



L'acide anthranilique lui même peut être obtenu en effectuant la réaction de dégradation d'Hoffmann de l'acide phtalique.