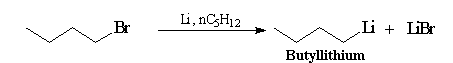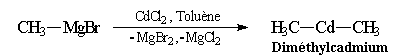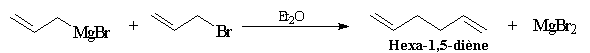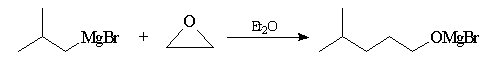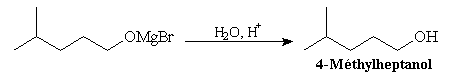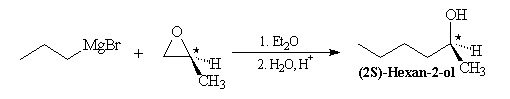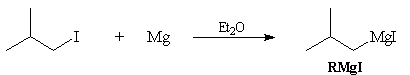
Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
LES
ORGANOMÉTALLIQUESIntroduction
Origine
Polarité de la liaison métal-carbone des organométalliques s
Le tableau ci-dessous permet de classer les organométalliques selon le
pourcentage de caractère ionique de la liaison C- métal. Dans tous les cas le
carbone porte une charge négative. Les organométalliques sont des carbanions
potentiels. L'importance de la charge négative qui se développe sur l'atome de
carbone dépend de l'électronégativité du métal et dans une certaine mesure
du solvant. On peut prévoir pour ces composés une réactivité basique
et nucléophile.
Schématiquement, la réactivité des carbanions décroît avec le pourcentage de caractère ionique de la liaison. Donc (en gros) des organopotassiques aux organocuivreux.
|
Élément |
K |
Na |
Li |
Mg |
Al |
Zn |
Cd |
Pb |
Hg |
Cu |
|
Electronégativité |
0,82 |
0,93 |
0,98 |
1,31 |
1,61 |
1,65 |
1,69 |
1,87 |
2,00 |
2,5 |
|
% Caractère ionique |
51 |
47 |
43 |
35 |
22 |
18 |
15 |
12 |
9 |
0 |
On peut schématiser la représentation de la liaison entre le carbone et le métal par les formes mésomères suivantes :
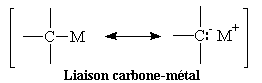
La liaison C- Mg possède un caractère ionique de 35 %. Cela confère aux magnésiens une position intermédiaire entre les organopotassiques et les organocuivreux.
Si l'on se réfère au dérivé halogéné R-X de départ, la polarité de la liaison entre le carbone et la partie inorganique a été inversée. Il s'agit d'un exemple d'inversion de la polarité d'une liaison. Ce concept, convenablement généralisé est souvent mis à profit en synthèse organique.
Préparations
Organosodiques
Les composés dans lesquels la charge négative est stabilisée par délocalisation peuvent être obtenus assez facilement par métallation de la liaison C-H de l'hydrocarbure correspondant.
Le fluorène (pKa = 23) est suffisamment acide pour pouvoir être déprotoné par l'ion hydroxyde dans le DMSO.
|
Le DMSO est un solvant dipolaire aprotique de permittivité relative élevée ( er = 49) entraînant une bonne dissociation des paires d'ions. Son moment dipolaire élevé (m = 4,3 D) et une charge négative dégagée sur l'atome d'oxygène, lui permettent de solvater les ions Na+ de façon efficace. Dans ce solvant, le caractère basique de l'ion OH- est fortement exalté. Le DMSO traverse très facilement la peau. Il faut porter des gants pour le manipuler. |
|
|
Un mélange d'hydroxyde de sodium en perles et de fluorène sont ajoutés à une petite quantité de DMSO bien sec. Le mélange est agité à la température ambiante. Au bout d'une quinzaine de minutes, on obtient une solution de couleur rouge qui contient l'anion fluorényle. Cet ion est stabilisé par résonance. La conjugaison est suffisamment étendue pour qu'il absorbe dans la partie visible du spectre. |
Les ions alcynyles sont aisément synthétisés par réaction entre un alcyne terminal (souvent appelé alcyne vrai) et une base comme l'ion amidure dans NH3 (l). L'acétylène (éthyne) pKa = 25 est un acide plus fort que l'ammoniac pKa = 33. Ainsi l'ion éthynyle peut être préparé en utilisant comme base l'ion amidure dans l'ammoniac liquide.
![]()
Les ions alcynyles sont d'excellents nucléophiles.
Préparation des organomagnésiens
R-X + Mg = RMgX
X désigne I, Br, Cl. La synthèse des organomagnésiens fluorés nécessite une procédure spécifique.
Mode opératoire
Le magnésium en tournures, conservé à l'étuve, est introduit dans le
ballon puis immédiatement recouvert d'éther anhydre. Une petite quantité du dérivé
halogéné contenu dans l'ampoule est introduit dans le ballon. Après une
courte période d'induction, la réaction démarre. De petites bulles traduisent
l'ébullition du solvant.
On peut accroître la vitesse de départ en élevant la température avec un bain-marie. La réaction étant exothermique elle est rapidement auto-entretenue à la température de reflux du solvant.
Lorsque la réaction a démarré, le dérivé halogéné introduit goutte à goutte à partir de l'ampoule de coulée. Cette manière de procéder permet de limiter autant que possible les réactions de couplage. Le magnésium disparaît complètement en fin de réaction si les réactifs ont été introduits en proportions stœchiométriques. Avec du magnésium ultra pur, la solution est incolore. Le plus souvent on obtient un mélange gris foncé à cause des impuretés du métal.
La réactivité des halogénures d'alkyles décroît dans le même ordre que la mobilité de la liaison carbone- halogène :
RI > RBr > RCl >> RF
Les bromures d'alkyle ont une réactivité un peu plus faible que celle des iodures. On préfère généralement utiliser les bromures en raison de leur coût moins élevé sauf dans le cas de CH3I qui présente l'avantage d'être le seul dérivé méthylé liquide à la température ordinaire.
Préparation des organolithiens
Les organolithiens peuvent être obtenus par réaction
entre le métal et un dérivé halogéné. Compte-tenu de la grande réactivité
de ces composés il est indispensable d'utiliser des réactifs rigoureusement
anhydres et de travailler en l'absence d'oxygène. On utilise un montage du même
type que le précédent et on procède à un balayage soigneux du dispositif par
un gaz inerte. Différents solvants peuvent être utilisés parmi lesquels : les
éthers, le pentane, le cyclohexane.
Le butyllithium est un réactif utile qu'on peut préparer par la réaction suivante :
|
|
On le trouve dans le commerce, sous forme de solution 2 mol.L-1 dans l'hexane ou l'éther. On l'utilise surtout en tant que base très forte pour générer des carbanions ou des amidures comme le LDA.
Les dérivés aromatiques peuvent être synthétisés par réaction directe mais ils sont obtenus de façon plus pratique par une réaction d'échange métal-halogène. Pour utiliser ces réactifs dans des conditions optimales quand le solvant est l'éther, il est préférable d'effectuer leur dosage avant réaction car les lithiens attaquent lentement le solvant. L'organométallique provoque une réaction d'élimination en arrachant un d'hydrogène situé sur un atome de carbone en b de l'oxygène.
Organocadmiens & organozinciques
|
|
Le magnésium étant plus électropositif (moins électronégatif) que le cadmium et le zinc, l'état final dans lequel le Mg est sous forme de sel et le cadmium dans l'organométallique est plus stable que l'état initial. Cette propriété est générale, dans ce type de réaction, l'élément métallique le plus électropositif se retrouve sous forme de sel. Les organocadmiens permettent la synthèse de cétones à partir des chlorures d'acyles.
Organocuprates
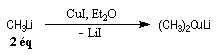
Ils permettent des additions 1, 4 très fortement régiosélectives sur les a-énones (addition de Michaël).
Nomenclature
|
C2H5MgCl |
PhLi |
(CH3)2 Cd |
(CH3)2CuLi |
|
Chlorure d’éthylmagnésium |
Phényllithium |
Diméthylcadmium |
Diméthylcuprate de lithium |
Structure, rôle du solvant
Généralités
Les éthers sont parmi les solvants les plus couramment utilisés mais on peut aussi utiliser des amines tertiaires comme la triéthylamine. Avec les magnésiens, si le solvant est très basique, la réactivité diminue.
Le choix du solvant dépend notamment de la réactivité de l'halogénure. Avec les dérivés halogénés ordinaires, on utilise l'éthoxyéthane. Avec les composés vinyliques, peu réactifs et benzéniques qui le sont tout de même davantage, on utilise le THF plus basique que l'éthoxyéthane et qui permet un reflux à température plus élevée (la difficulté est parfois de disposer de THF rigoureusement anhydre).
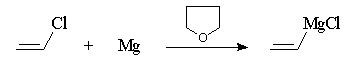
L'utilisation des magnésiens vinyliques dans le THF permet d'introduire une double liaison dans une chaîne carbonée.
Structure à l'état solide
La présence d'un solvant donneur d'électrons est essentielle pour synthétiser
un organomagnésien. Avec les éthers, on a pu mettre dans certains cas en évidence
à l'état solide, la présence de deux molécules d'éther par molécule
d'organométallique.
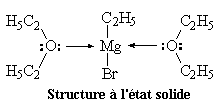
Il existe des structures plus complexes qui font intervenir deux molécules d'organomagnésien.
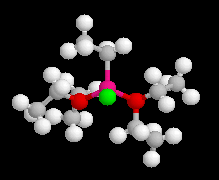
|
|
La structure d'un certain nombre de composés organomagnésiens à l'état solide a été déterminée par diffraction des rayons X. Dans [EtMgBr(OEt2)2] le magnésium est au centre d'un tétraèdre irrégulier. Sur l'image de gauche l'atome de magnésium est représenté en rose, celui de brome en vert et l'atome d'oxygène en rouge. |
Structure en solution
En solution, la situation est plus compliquée. Plusieurs équilibres
interviennent simultanément et leur position dépend de plusieurs facteurs
(concentration, température, solvant, etc.)
On peut mettre en évidence un équilibre entre magnésien mixte et symétrique. Dans le dioxane cet équilibre est déplacé en faveur du magnésien symétrique car dans ce solvant, MgX2 qui est peu soluble, précipite.
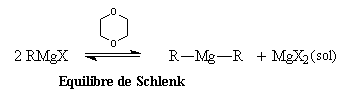
Le déplacement de l'équilibre de Schlenk dans le dioxane favorise les réactions de réduction des cétones dans le cas de magnésiens encombrés possèdant un atome d'hydrogène sur le carbone situé en b du magnésium.
Mécanisme de formation des organomagnésiens
Bien qu'il ait fait l'objet d'un nombre très important d'études, le mécanisme
de formation des organomagnésiens n'est pas connu avec certitude. Globalement
il y a eu insertion de magnésium dans la liaison carbone- halogène. Il est
vraisemblable que le mécanisme implique une réaction en chaîne avec
intervention de radicaux.
Réactivité basique
Généralités
Sont déprotonés de façon quantitative les fonctions suivantes : acides carboxyliques, phénols, alcools, amines, amides non substitués, alcynes terminaux. Certains H acides portés par des atomes de carbone en a de fonctions carbonyles peuvent aussi être arrachés, notamment dans le cas des composés dicarbonylés qui possèdent des atomes d'hydrogène d'acidité notable.
Avec les magnésiens la réaction bilan s'écrit : R-Mg-X + A-H = R-H + MgXA
La réaction avec l'eau s'écrit souvent de façon simplifiée :
|
|
En réalité, les magnésiens étant fortement basiques, l'hydrolyse conduit à la formation de Mg(OH)2 (s) qui se présente sous la forme d'un solide blanc.
|
|
|
|
L'ajout de quelques gouttes d'organomagnésien à une solution hydroalcoolique de phénolphtaléine entraîne un changement de couleur de l'indicateur en même temps, on observe la précipitation de Mg(OH)2. Si la quantité de magnésien est importante la solution se décolore car le milieu est devenu tellement basique que la phénolphtaléine passe sous forme incolore. |
Afin d'éviter la précipitation de Mg(OH)2 , on réalise le plus souvent l'hydrolyse des magnésiens en milieu acide avec une solution de NH4Cl ou d'acide sulfurique dilué.
|
|
Ces réactions constituent le plus souvent une limitation à l'emploi des organomagnésiens. On peut aussi les mettre à profit dans quelques cas particuliers.
Dosage des organomagnésiens & des organolithiens
La réaction entre un organomagnésien ou un organolithien et un alcool
comme le butanol est quantitative. Cette réaction peut être donc mise à
profit pour effectuer le dosage de l'organométallique.
|
|
|
Méthode de Zérévitinov
Cette méthode est assez ancienne. L'organomagnésien est mis à réagir
avec une solution de concentration connue d'un composé à hydrogène mobile
dissous dans un solvant inerte. On obtient un alcane dont on mesure le volume.
Inversement, avec une solution titrée d'organomagnésien, on peut doser des
composés à hydrogène mobile.
Synthèse des magnésiens alcyniques
Les alcynes étant beaucoup plus acides que les alcanes, l'atome d'hydrogène
d'un alcyne terminal est arraché par l'organométallique. On a ainsi une voie
d'accès aux magnésiens alcyniques qu'on ne peut pas préparer par union
directe entre un halogénure et le magnésium.
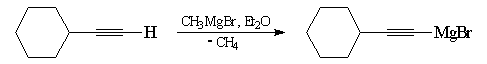 .
.
Les magnésiens dérivant des alcynes sont des réactifs utiles qui permettent l'allongement des chaînes carbonées tout en préservant la possibilité de réactions d'addition sur la liaison triple.
Réactions de substitutions nucléophiles
Généralités
Les réactions de couplage interviennent notamment avec les iodures d'alkyles en raison de la grande polarisabilité de la liaison carbone- iode.
Ces réactions sont souvent des réactions parasites dans la préparation des magnésiens et des lithiens. On les limite en ajoutant le dérivé halogéné goutte à goutte et en travaillant le métal en excès.
Halogénures allyliques, synthèses de diènes
La préparation de diènes 1,5 peut être effectuée à partir des
halogénures allyliques qui sont très réactifs (une manifestation de la
mobilité des liaisons carbone- halogène en position allylique).
|
|
Les magnésiens allyliques déplacent facilement l'halogène du substrat. C'est la raison pour laquelle on doit utiliser des solutions diluées d'halogénure et le métal en grand excès pour préparer ces organométalliques.
|
|
Dans ce cas le mécanisme s'apparente à une substitution SN1 avec formation d'un carbocation allylique.
Alkylation des ions alcynyles
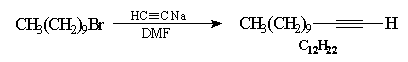
L'alkylation des ions alcynyles est une méthode intéressante d'introduction d'une liaison triple dans une chaîne carbonée. Les liaisons triples peuvent selon les, cas être transformées en doubles liaisons de configuration Z ou E bien définie : Z par addition de H2 en présence de Pd Lindlar ou E par réaction avec Na dans NH3 (l).
Alkylation des alcénylcuprates
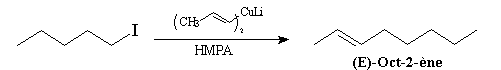
La stéréochimie de l'alcène est définie par celle du cuprate.
Ouverture des époxydes
Définitions
Les époxydes
sont des éthers cycliques à trois chaînons. Ces composés tendus, très réactifs
peuvent être ouverts sous l'action d'une grande variété de réactifs nucléophiles.
Globalement la réaction entre l'organométallique et l'époxyde est une
addition. Sur le plan du mécanisme, on peut la regarder comme une réaction de
substitution nucléophile, dans laquelle l'organométallique est le réactif et
l'époxyde est le substrat.
Organomagnésiens et organolithiens
La réaction s'effectue en deux étapes distinctes : l'addition du réactif
organométallique à l'époxyde conduit à un alcoolate. La réaction entre un
organomagnésien et l'oxyde
d'éthylène permet d'allonger une chaîne carbonée de
deux atomes de carbone.
|
|
Cet alcoolate magnésien est hydrolysé en milieu acide (NH4Cl ou H2SO4 2 M) afin d'éviter la précipitation de Mg(OH)2.
|
|
L'organomagnésien réagit du côté opposé au pont et la réaction procède d'un mécanisme de type SN2. Lorsque l’époxyde est dissymétrique la réaction préférentielle implique l'atome de carbone qui est stériquement le plus dégagé. Conformément au mécanisme précédent, la réaction s'accompagne en général d'une inversion de configuration de cet atome.
|
|
Réactions d'additions nucléophiles
Carbonylés : aldéhydes et cétones
Aldéhydes
Les aldéhydes sont plus réactifs que les cétones. La réaction entre un
organomagnésien et un aldéhyde, suivie d'une hydrolyse acide, constitue une
synthèse d'alcool secondaire.
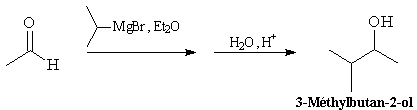
Lorsque l'aldéhyde est le méthanal, on obtient un alcool primaire.
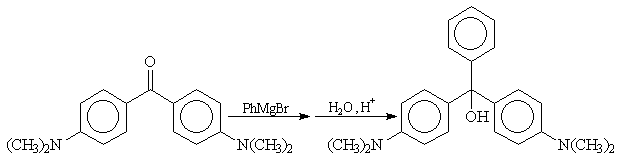
Cétones
La réaction entre un organomagnésien et une cétone conduit à un alcool
tertiaire.
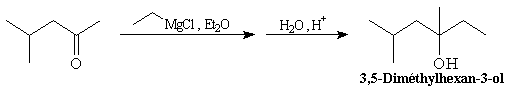
|
|
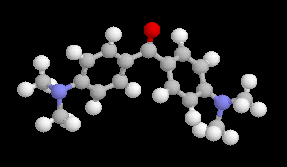
L'image de gauche représente la cétone de Michler encore appelée : 4,4'-bis(diméthylamino)- benzophénone en nomenclature officielle. Ce composé est utilisé comme réactif de base dans l'industrie des colorants. |
La réaction entre le bromure de phénylmagnésium et la cétone de Michler conduit à un alcoolate magnésien. Après hydrolyse en milieu acide on obtient l'alcool tertiaire.
|
|
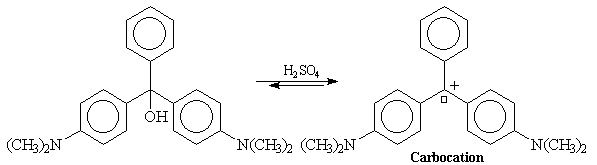
En milieu acide, il s'établit un équilibre entre cet alcool et le carbocation par perte d'eau. La force motrice de cette réaction est la formation d'un système fortement conjugué impliquant trois cycles aromatiques.
|
|
La conjugaison est suffisamment étendue pour que le système absorbe la lumière dans le domaine visible. Le carbocation formé confère à la solution une belle couleur verte.
|
|
L'erlenmeyer contient une solution de bromure de phénylmagnésium dans l'éthoxyéthane. On ajoute successivement 1 mL d'une solution de cétone de Michler dans le toluène, 1 mL d'eau distillée puis quelques gouttes d'acide acétique. Après agitation, une couleur bleu vert de développe. Le carbocation obtenu est utilisé comme colorant. |
Possibilités synthétiques
|
Composé carbonylé |
Classe de l’alcool formé |
|
Méthanal |
Primaire |
|
Aldéhyde (à part méthanal) |
Secondaire |
|
Cétone |
Tertiaire |
Il existe généralement plusieurs choix possibles de substrat et de réactif organométallique pour synthétiser une molécule cible donnée. Le choix d'un couple particulier doit tenir compte des réactions concurrentes d'énolisation et de réduction.
Énolisation
Dans le cas de substrats encombrés, l'énolisation du substrat par
l'organomagnésien jouant le rôle de base, peut devenir plus rapide que
l'addition nucléophile sur le carbonyle.
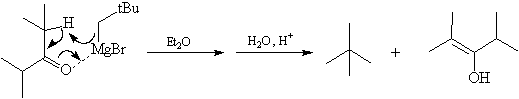
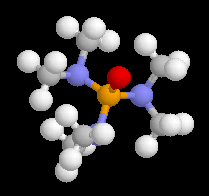
L'énol se tautomérise rapidement pour redonner la cétone de départ. Le pourcentage d'énolisation dépend du solvant utilisé. Dans des solvants très solvatants comme HMPT le pourcentage d'énolisation peut devenir très élevé.
|
|
L'hexaméthylphosphotriamide (HMPT) possède une constante diélectrique assez élevée ( er = 30) et surtout .un moment dipolaire très grand (m = 5,5 D). La charge négative est relativement dégagée tandis que la charge positive portée par le phosphore est masquée et dispersée par les substituants (observer le modèle "spacefill"). HMPT possède un pouvoir solvatant important vis à vis des sites déficients en électrons du magnésium et la liaison carbone- métal des magnésiens est fortement polarisée. Dans ce solvant, les magnésiens ont une action énolisante vis à vis des composés carbonylés. |
On peut mettre ces propriétés à profit pour synthétiser des cétones à partir des esters.
Réduction
Cette réaction intervient spécialement lorsque l'organomagnésien possède
un atome d'hydrogène sur l'atome de carbone en b .
La réaction entre la 4,4-diméthylpentan-2-one et le bromure de 2-méthyléthylemagnésium suivie d'hydrolyse, fournit trois produits. A côté de l'alcool tertiaire résultant de l'addition de l'organomagnésien sur la cétone, on obtient un alcool secondaire et du méthylpropène.
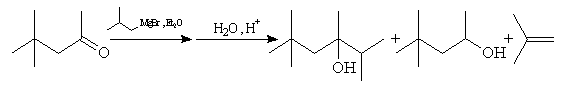
On interprète cette réaction par l'addition nucléophile d'un ion hydrure de l'organomagnésien sur le groupe carbonyle de la cétone.
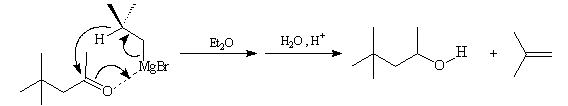
Elle rappelle la réduction des carbonylés par l'isopropylate d'aluminium (réaction de Meerwein- Pondorff et Verley). Cette réaction est favorisée dans le dioxane qui permet de déplacer l'équilibre de Schlenk vers la formation du magnésien symétrique. Dans ce solvant, R2Mg interagit avec l'oxygène du carbonyle de la cétone et favorise le transfert d'ion hydrure.
La synthèse des alcools encombrés peut être réalisée en utilisant des organolithiens. Ceux-ci étant beaucoup plus réactifs que les organomagnésiens, l'addition nucléophile peut être effectuée à basse température (-70 °C) ce qui minimise les réactions précédentes.
Mécanismes
Les mécanismes de réaction entre les organomagnésiens et les composés
carbonylés ne sont pas complètement élucidés. Dans certains cas, la cinétique
de la réaction est en faveur d'un mécanisme dans lequel intervient deux molécules
d'organomagnésien (mais une loi cinétique ne permet pas de démontrer un mécanisme).
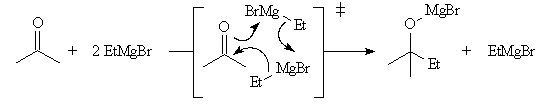
La réaction entre les organomagnésiens et les cétones aromatiques a été particulièrement étudiée par Ashby qui a pu démontrer l'intervention d'un mécanisme radicalaire.
a- énonesIntroduction
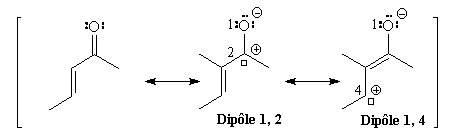
Réactivité
Un magnésien peut réagir directement avec le groupe carbonyle (addition 1, 2)
ou bien réagir avec l'atome de carbone en b du
groupe carbonyle (addition 1, 4). L'hydrolyse de ce dernier composé fournit l'énol
qui se tautomérise rapidement en cétone.
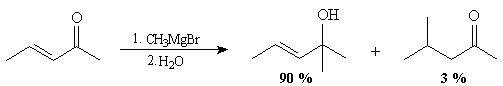
En présence de sels cuivreux comme CuBr, l'addition 1-4 des magnésiens est favorisée.
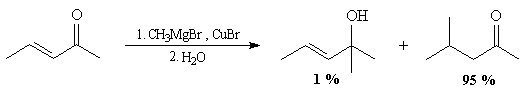
Les lithiens réagissent de façon préférentielle en 1,2.
Les cuprates lithiens tels que Li(CH3)2Cu réagissent en 1, 4 de façon régiospécifique. L'organométallique réagit sur l'atome de carbone 4, là où le coefficient de l'orbitale atomique est le plus grand.
Fonctions dérivées d'acides
Réactivité
Schématiquement, on doit tenir compte de deux facteurs :
|
Caractère électrophile du carbone |
Aptitude nucléofuge du groupe partant |
|
RCOCl > RCOOR' > RCONHR2 |
Cl- > RO- >> NR2- |
Chlorures d'acyle
L'intermédiaire tétraédrique issu de l'addition nucléophile du magnésien
sur le carbonyle est thermodynamiquement instable vis à vis de sa
transformation en cétone et labile du fait du caractère nucléofuge de l'ion
chlorure. Le magnésien réagit sur la cétone formée et le produit final, à
la température ordinaire, est généralement l'alcool tertiaire. Il est
cependant possible de synthétiser une cétone en mettant à profit la plus
grande réactivité des chlorures d'acyles :
Lorsque la température est suffisamment basse (-100 °C) en utilisant le THF comme solvant, le chlorure d'acyle réagit plus vite que la cétone vis à vis de l'organomagnésien. Celle-ci n'est donc pas transformée en alcool tertiaire.
Le remplacement de l'organomagnésien par un organocadmien permet de s'arrêter au stade cétone. En effet les cétones moins réactives que les chlorures d'acyles ne réagissent pas avec les cadmiens. Cela est illustré par la réaction suivante :
![]()
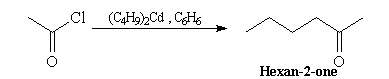
Esters
La réaction est comparable à la précédente Les esters ont une réactivité
moins grande que celle des chlorures d'acyles. Le produit d'addition initial
entre le magnésien et l'ester est un intermédiaire tétraédrique (I).
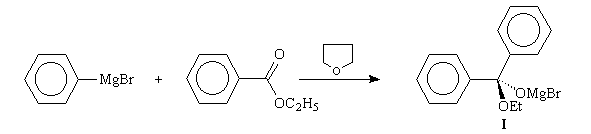
Cet intermédiaire est labile car l'ion alcoolate, sans être un excellent nucléofuge l'est suffisamment pour que l'élimination soit effective à la température du reflux du solvant. L'intermédiaire évolue rapidement dans le milieu réactionnel en cétone.
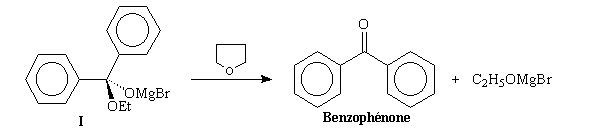
Puisque les cétones réagissent généralement plus rapidement que les esters on obtient la plupart du temps un alcool tertiaire dans lequel 2 groupes sont identiques. Cela est illustré par la synthèse du triphénylméthanol.
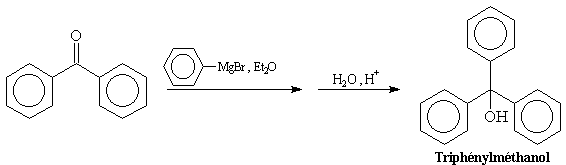
Il existe cependant des synthèses de cétones à partir des esters dans quelques cas particuliers :
Dans le cas où la cétone formée est très encombrée, l'addition du magnésien sur le carbonyle est fortement ralentie et on peut isoler la cétone. La réaction entre un organomagnésien et un ester de l’acide méthanoïque (formique) HCOOH, constitue une synthèse d'alcool secondaire.
On peut mettre à profit l'action énolisante des magnésiens dans l'HMTP. Dans ce solvant très solvatant, la cétone intermédiaire est énolisée ce qui bloque la réaction d'addition de l'organomagnésien.
Amides
Le produit d'addition du magnésien sur l'amide est thermodynamiquement
instable par rapport au composé carbonylé (à cause de la grande énergie de
liaison du carbonyle) mais cinétiquement inerte car NR2-
est un mauvais nucléofuge.
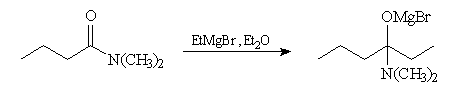
Le passage en milieu acide entraîne l'élimination de R2NH2+ à partir de l'intermédiaire. Ce nucléofuge est en effet bien meilleur que NR2- (c'est une base beaucoup plus faible).
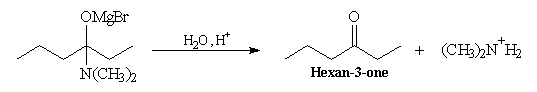
Il est impératif d'utiliser un amide N,N-disubstitué dans cette réaction. Avec un amide non substitué, le magnésien arrache un atome d'hydrogène à l'amide et est détruit.
Nitriles
L'addition du magnésien au nitrile conduit à la formation d'un sel d'immonium.
Ce sel d'imine bien qu'insaturé ne réagit pas avec le magnésien car cela
impliquerait deux charges négatives sur le même atome d'azote.
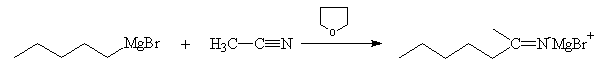
L'hydrolyse du sel d'immonium le transforme en iminoalcool. En milieu acide, ce dernier fournit la cétone plus stable.
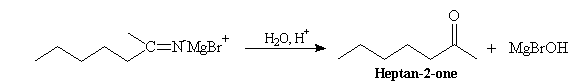
Cette réaction qui constitue une très bonne méthode de synthèse des cétones possède une limitation dans le cas ou l'atome d'hydrogène situé sur l'atome de carbone en a de la fonction nitrile est trop acide. Il y a alors énolisation.
Réaction avec le dioxyde de carbone
Le dioxyde de carbone peut être considéré comme l'anhydride de l'acide
carbonique. Sa réaction avec un organomagnésien
constitue une excellente méthode de synthèse des acides carboxyliques.
L'addition de l'organomagnésien sur l'atome de carbone électrophile de CO2
conduit à un composé qui comporte encore une insaturation. Bien que l'addition
d'une deuxième molécule de magnésien sur cet intermédiaire soit généralement
beaucoup plus lente que la première, on peut s'en affranchir complètement en
versant le magnésien sur de la carboglace.
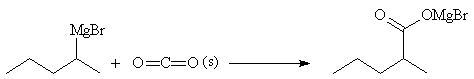
Après acidification, on obtient l'acide.
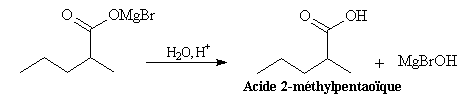
La réaction avec les lithiens est tout à fait comparable mais ceux-ci étant plus réactifs que les magnésiens la réaction avec le sel intermédiaire devient importante si le lithien est en excès. On obtient alors après hydrolyse l'alcool tertiaire.
La carbonatation des magnésiens dérivant de composés aromatiques est une méthode intéressante d'introduction d'un groupe carboxyle sur un cycle aromatique.
Corps simples
Dioxygène
La réaction entre un organomagnésien et le dioxygène en excès à basse température
(mélange carboglace- acétone) suivie d'hydrolyse, est une très bonne méthode
de préparation des hydroperoxydes.
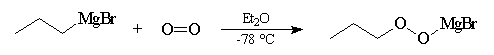
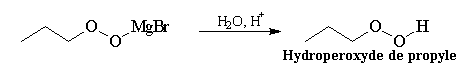
Le contrôle de la température est important dans cette synthèse car les peroxydes organiques sont des composés peu stables en raison de la faible énergie de dissociation de la liaison peroxo qui lie les deux atomes d'oxygène. Ils sont potentiellement explosifs et donc dangereux.
Si l'organomagnésien est en excès, il réagit avec le peroxyde d'halogénure magnésium et on obtient l'alcool après hydrolyse.
Soufre
La réaction s'effectue par union directe entre le magnésien et le soufre.
Après hydrolyse, on obtient un thiol ou un thiophénol qu'on peut considérer
respectivement comme les analogues soufrés des alcools ou des phénols.
Dihalogènes
On peut regarder cette réaction comme une substitution nucléophile. Dans
ce cas R- MgX est le réactif et X2
le substrat.
R- MgX + X2 = R- X + MgX2
La réaction avec I2 permet d'effectuer le dosage des organomagnésiens. On introduit I2 en excès. On dose I2 restant par le thiosulfate.